
This Key Event Relationship is licensed under the Creative Commons BY-SA license. This license allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use. If you remix, adapt, or build upon the material, you must license the modified material under identical terms.
Relationship: 908
Title
Increase, Mitochondrial dysfunction leads to Degeneration of dopaminergic neurons of the nigrostriatal pathway
Upstream event
Downstream event
Key Event Relationship Overview
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding | Point of Contact | Author Status | OECD Status |
|---|---|---|---|---|---|---|
| Inhibition of the mitochondrial complex I of nigro-striatal neurons leads to parkinsonian motor deficits | non-adjacent | Moderate | Low | Andrea Terron (send email) | Open for citation & comment | WPHA/WNT Endorsed |
| Inhibition of the mitochondrial complex III of nigro-striatal neurons leads to parkinsonian motor deficits | adjacent | Barbara Viviani (send email) | Under development: Not open for comment. Do not cite | |||
| Inhibition of the mitochondrial complex II of nigro-striatal neurons leads to parkinsonian motor deficits | adjacent | Barbara Viviani (send email) | Under development: Not open for comment. Do not cite | |||
| Inhibition of the mitochondrial complex IV of nigro-striatal neurons leads to parkinsonian motor deficits | adjacent | Barbara Viviani (send email) | Under development: Not open for comment. Do not cite |
Taxonomic Applicability
Sex Applicability
| Sex | Evidence |
|---|---|
| Male | High |
| Female | High |
Life Stage Applicability
| Term | Evidence |
|---|---|
| All life stages | High |
Key Event Relationship Description
Neurons are characterized by the presence of neurites, the formation of action potentials, and the release and re-uptake of neurotransmitters into the synaptic cleft. The presence of long extensions implies a significant enlargement of total cell surface. In combination with the transmission of action potentials that require a continuous maintenance of active transport processes across the membrane, the steady state energy demand of these neurons is significantly higher compared with non-neuronal cells. Dopaminergic (DA) neurons located in the substantia nigra pars compacta (SNpc) that project into the striatum are unique with respect of the total length of their neurites and the number of synapses that are significantly higher compared with other neuronal cell types (Bolam et al., 2012). Besides this complex morphology DA neurons have a distinctive physiological phenotype that could contribute to their vulnerability (Surmeier et al., 2010). Other features such as high energy demand, high calcium flux, dopamine autoxidation process as well as high content of iron and high content of microglia makes these DA neurons at vulnerable population of cells to oxidative stress produced by mitochondrial dysfunction. These architectural features of SNpc DA neurons render this cell type as particularly vulnerable to impairments in energy supply. Mitochondrial dysfunction, either evoked by environmental toxins such as the complex I inhibitor rotenone or MPTP, by oxidative modifications of components of the mitochondrial respiratory chain, or by genetic impairments of mitochondrial ATP generation hence have direct influence on the function and integrity of SNpc DA neurons.
Evidence Collection Strategy
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02): The implementation of AOP3 is based on a negotiated procedure with EFSA (reference NP/EFSA/PREV/2024/02). This procedure is intended to update AOP3 by adding more evidence to the AOP Wiki, considering the contribution of mitochondrial complex I and III inhibition, for which a strong biological plausibility that their inhibition lead to degeneration of dopaminergic neurons exist. The starting conceptual model for this project is based on the key scientific sources, including EFSA (2017), Delp et al. (2019 and 2021), Van der Stel et al. (2020 and 2022), ENV/JM/MONO(2020)22, Alimohammadi et al. (2023), Tebby et al (2022). These publications provided the initial basis for this project and contributed to the Empirical Evidence. Additional literature was identified through a structured, non systematic search using a stressor-based search strategy as described in the “AOP development strategy” section. - Not endorsed
Evidence Supporting this KER
Biological Plausibility
Mitochondria are organelles essentials for multiple cellular processes, including production of ATP, maintenance of calcium homeostasis, management of ROS production and apoptosis. Mitochondrial dynamics are also critical for the maintenance of cellular homeostasis, which involve multiple factors controlling mitophagy (Youle et al. 2012). Deregulation of mitochondrial functions may impact any neuronal population; however, SNpc DA neurons are indeed the most vulnerable population in PD. Multiple factors are related to their vulnerability: These include autonomous activity, broad action potentials, low intrinsic calcium buffering capacity, poorly myelinated long highly branched axons and terminal fields, and use of a catecholamine neurotransmitter, often with the catecholamine-derived neuromelanin pigment (Sulzer et al. 2013; Surmeier et al.2010).
The above mentioned factors imply a significantly higher total cell surface and a high energy requirement in order to maintain the re-distribution of ions across the membrane following an action potential. In addition, SNpc DA neurons are characterized by significantly higher numbers of synapses compared with other neuronal types or with DA neurons of different anatomical localizations (Anden et al., 1966; Kawaguchi et al., 1990; Kita et al., 1994; Bevan et al., 1998; Wu et al., 2000; Tepper et al., 2004). In humans, ca. 10 times higher numbers of synapses compared with rats are expected, making human DA neurons particularly vulnerable (Bolam et al., 2012; Matsuda et al., 2009). These extreme bioenergetics demands pose SNpc DA neurons energetically “on the edge”. Any stressor that might perturb energy production would hence lead to conditions under which the energy demand would exceed energy supply, resulting in cell damage and ultimately to cell death.
The mechanistic link between mitochondrial dysfunction and loss of SNpc DA neurons also comes from evidence of mutated proteins related to mitochondrial function in familial PD, resulting in reduced calcium capacity, increased ROS production, increase in mitochondrial membrane permeabilization and increase in cell vulnerability (Koopman et al. 2012; Gandhi et al. 2009). In addition, excessive ROS production can damage mitochondrial DNA and activate the intrinsic pathway of apoptosis (Tait et al. 2010). Additional sources of oxidative stress come from the autoxidation of dopamine and the active generation of ROS by activated glia cells; furthermore, the mitochondrial respiratory chain itself represents a source of constant superoxide formation, even under normal conditions (Moosmann et al., 2002).
Imbalance of mitochondrial dynamics have been also reported in a wide range of experimental models of PD and inhibition of the mitochondrial fission proteins (i.e. Drp1) promote mitochondrial fusion and fission and enhanced the release of dopamine from the nigrostriatal terminals (Tieu et al. 2014).
Additional link between mitochondrial dysfunction and the degeneration of DA neurons of the nigrostriatal pathway comes from studies indicating a reduced activity of mitochondrial complex I in human idiopathic PD cases in the substantia nigra (Keeney et al., 2006; Parker et al., 1989, 2008; Swerdlow et al., 1996). The impairment in complex I activity was directly correlated with an elevated sensitivity of SNpc DA neurons and their demise. Transfer of mitochondria from human platelets collected from idiopathic PD subjects into fibroblasts or neuronal cells resulted in elevated levels of basal oxidative stress, a declined supply with ATP, and an elevated vulnerability towards exogenous stressors such as the complex I inhibitors rotenone or the redox cycler paraquat (Swerdlow et al., 1996; Gu et al., 1998). Systemic application of complex I inhibitors such as rotenone or MPTP lead to a preferential loss of nigrostriatal DA neurons, while other brain areas or peripheral cells are not affected to the same degree (Langston et al., 1983).
Empirical Evidence
The experimental support linking mitochondrial dysfunction with the degeneration of DA neurons of the nigrostriatal system is based on the analysis of mitochondria from PD patients, from genetic mouse models, from in vitro knockdown and overexpression systems, and from in vitro and in vivo toxin models.
- In vitro/rotenone: Prevention of ROS formation protects from cell death. The concept of mitochondrial dysfunction as a consequence of defects in complex I has been fueled by observations of impaired complex I activity in the SNpc, muscle, and in platelets of PD patients. Human neuroblastoma SK-N-MC cells, exposed to rotenone, displayed a time- and concentration-dependent decline in viability. Transfection of rotenone-insensitive single subunit NADH dehydrogenase (NDI 1) allowed a replacement of endogenous complex I activity. NDI 1 transfected cells showed no oxidative damage, no declined mitochondrial activity, or cell death. A significant amount of endogenously formed ROS at complex I was identified in SK-N-MC cells and in a chronic midbrain slice culture exposed to rotenone. Antioxidants such as -tocopherol prevented cell death evoked by rotenone, but not the rotenone-induced drop in ATP (Sherer et al. 2003).
- In vitro/rotenone/MPP+: Antioxidants protect from rotenone/MPP+ cell death. Analysis of post mortem nigrostriatal material from PD patients regularly revealed the presence of elevated levels of oxidative modified proteins, lipids, and DNA. These observations indicate an elevated formation of ROS in the cells affected by the disease and triggerd the concept of antioxidants as a potential intervention strategy to slow down the progression of PD. In MES23.5 cells, a reduction in viability, DA content, NADH levels, as well as an increase in ROS formation and elevated nuclear condensation was observed upon treatment with MPP+. Rosmarinic acid is well known for its radical scavenging activities and displayed a complete protection from MPP+-mediated cell death and rescued NADH levels. In addition, it lead to a partial protection from the loss of DA and resulted in a rate of nuclear condensation that was about half of that observed with MPP+ alone (Du et al. 2010). The flavonoid rutin has been demonstrated to protect from oxidative stress in 6-OHDA induced motor deficits in rats as well as to inhibit the formation of nitric oxide and proinflammatory cytokines (Khan et al., 2012). In a model of SH-SY5Y cells, exposure to rotenone lead to a reduction in viability by ca. 50% that was almost completely protected in the presence of rutin. Rotenone-dependent increase of ROS formation and an elevation of intracellular Ca2+ was significantly dampened by the presence of rutin, similar to its rescue from rotenone-dependent decrease in mitochondrial membrane potential (Park et al., 2014). Comparable observations were made with the quinone triterpene celastrol that protected SH-SY5Y cells exposed to rotenone almost completely from cell death, from a rotenone-dependent elevation in ROS levels, and from a rotenone-dependent loss of the mitochondrial membrane potential (Choi et al., 2014).
- In vitro/different complex I inhibitors: Inhibition of complex I triggers oxidant formation and cell death. The majority of experimental PD studies were either conducted using rotenone or MPP+. In order to demonstrate that the concept of complex I inhibition and its ROS-mediated triggering of mitochondrial dysfunction and cell demise can be regarded as a general principle, alternative complex I inhibitors were applied to substantiate previous observations made with rotenone. In human SK-N-MC neuroblastoma cells, rotenone as well as the pesticides fenazaquin, fenpyroximate, pyridaben, tebufenpyrad, pyridaben were tested. In all cases, a time- and concentration-dependent decline in intracellular ATP and cell viability was observed. Expression of the rotenone-insensitive NADH dehydrogenase from Saccharomyces cerevisiae (NDI 1) prevented from the toxicity of the different complex I inhibitors completely. Rotenone- and pyridaben-dependent cell death was prevented by ca. 75 % by the presence of the antioxidant α-tocopherol.(Sherer et al., 2007).
- In vitro/rotenone: Mitochondrial dysfunction-dependent cell death is prevented by antioxidants. In a human neuroblastoma SH-SY5Y model, exposed either to the complex I inhibitors MPP+ or rotenone, the imine antioxidants iminostilbene, phenothiazine, phenoxazine in the low nanomolar concentration range partially protected from MPP+ or rotenone toxicity. A reduction in the membrane potential evoked by MPP+ and rotenone was completely prevented by these antioxidants (Hajteva et al., 2009).
- In vitro/rotenone: Circumvention of dysfunctional mitochondria protects from cell death. Assuming a direct causal relationship between complex I inhibition, mitochondrial dysfunction, and the demise of DA neurons, the circumvention of endogenous complex I by expression of the NADH dehydrogenase of Saccharomyces cerevisiae (NDI 1) provided initial evidence for the essential role of complex I inhibition in this sequence of events. As an alternative electron carrier, capable of transferring electrons from NADH to cytochrome c, methylene blue was identified. In hippocampal HT-22 cells, a rotenone-mediated reduction in the oxygen consumption rate was completely reversed by the addition of methylene blue. A rotenone-mediated decline in cell viability by 70 % was almost completely prevented by 0.1 µg/ml methylene blue . In rats, rotenone-mediated decline in striatal DA was entirely prevented by methylene blue, the observed elevation of ROS formation evoked by rotenone was reduced to control levels, and rotarod performance impairments evoked by rotenone were completely avoided by administration of methylene blue. These observations illustrate a causal relationship between dysfunctional mitochondria, the degeneration of nigrostriatal DA neurons, and impaired motor performance (Wen et al. 2011).
- In vivo/rotenone: Circumvention of dysfunctional mitochondria prevents from nigrostriatal cell degeneration. Circumvention of a dysfunctional complex I by the rotenone-insensitive NADH dehydrogenase NDI 1 in vivo and its influence on nigrostriatal DA neuron integrity was demonstrated in a rat model with an unilateral injection of a recombinant adeno-associated virus, carrying the NDI 1 gene into close special vicinity to the SNpc. The animals were treated with rotenone after the unilateral expression of NDI 1. NDI 1 almost completely prevented from the rotenone-mediated loss of TH staining in the SNpc and the striatum. Striatal DA levels that were reduced by ca. 50 % by rotenone, in the presence of NDI 1, DA levels were also almost identical to the values of untreated controls. These observations highlight a causal relationship between the inhibition of complex I and the degeneration of nigrostriatal DA neurons (Marella et al. 2008).
- In vitro/DA: Exogenously added oxidants lead to mitochondrial dysfunction and cell death. Next to an elevated formation of reactive oxygen species evoked by endogenous defects in complex I or in response to pharmacological inhibitors of complex I, nigrostriatal DA neurons are characterized by the neurotransmitter dopamine and its tendency to undergo autoxidation when exposed to physiological pH and oxygen tension conditions. To assess the role of DA-mediated oxidative stress as a cause of mitochondrial dysfunction and its influence on cell viability, PC12 cells were exposed to DA. The observed increase in intracellular ROS was completely reversed by the presence of the antioxidant N-acetly-cysteine (NAC). The amount of oxidative modified protein increased by DA treatment, its rise was completely prevented by the presence of NAC, and partially prevented by the presence of exogenously added GSH. DA-dependent PC12 cell death, decline in the transmembrane potential and in intracellular ATP, and decline in complex II/III activities were observed and were all completely prevented by the presence of NAC. (Jana et al., 2011). - In vitro/ GSH depletion: Oxidative stress causes mitochondrial dysfunction and neurodegeneration. Several reports indicated a declined activity of complex I in the brain, but also in muscle and platelets of PD patients. In order to investigate the mutual interaction between pro-oxidative conditions and complex I activity, a PC12 subclone was generated, allowing the inducible downregulation of -glutamyl-cystein synthetase involved in the synthesis of glutathione (GSH). This system allows a controlled decrease of intracellular GSH by ca. 50 % and a decrease in mitochondrial GSH by ca. 40 %. Under these conditions, intracellular and intramitochondrial ROS increased by ca. one third, mitochondrial complex I activity and ATP levels were reduced by ca. two thirds. The observed inhibition of complex I was completely reversed by DTT. These observations indicate that an impairment of complex I activity as a key event in the initiation of mitochondrial dysfunction and ultimately cell death, can be evoked by elevated levels of oxidants, respectively by a declined cellular antioxidant capacity (Jha et al., 2000).
- In vitro/ GSH depletion: Oxidative stress causes mitochondrial dysfunction and neurodegeneration. PD is characterized by the depletion of glutathione (GSH) in the SNpc. Declined cellular levels of GSH were reported to be associated with morphological changes of mitochondria (Perry et al., 1982; Jain et al., 1991). To investigate the influence of declined GSH levels, N27 cells were exposed to buthionine-S-sulfoximine (BSO), an inhibitor of glutamate cysteine ligase and hence of de novo GSH synthesis. The BSO concentration chosen allowed a reduction in intracellular GSH levels by 50 % in the absence of cell death. Chronic GSH depletion resulted in the S-nitrosation of complex I and its inhibition. Both effects were completely reversed by the addition of DTT (Chinta et al., 2006). - Isolated mitochondria: Exogenous oxidants cause mitochondrial dysfunction. In order to further address the aspect on how DA autoxidation contributes to mitochondrial dysfunction and DA neurodegeneration, isolated rat brain mitochondria were exposed to DA, resulting in an inhibition of complex I by ca. 30 % and in an inhibition of complex IV by ca. 50 %. Both activities of complex I and complex IV were completely protected from DA-dependent inactivation by the presence of GSH. These observations point to a direct inhibitory action of endogenous DA and its autoxidation derivatives on the activity of the mitochondrial respiratory chain. (Khan et al., 2005) - In vitro/cybrid cells: Sensitization of neuronal cells for degeneration by transfer of dysfunctional mitochondria. In a subclone of human neuroblastoma cells (SH-SY5Y), devoid of mitochondrial DNA, mitochondria from platelets of PD patients were transplanted. Analysis after 5-6 weeks in culture after transplantation of mitochondria indicated a 20 % reduction in complex I activity, a 2-fold increase in the basal formation of reactive oxygen species, and a ca. 2-fold higher sensitivity towards the mitochondrial PD toxin MPP+ (Swerdlow et al., 1996).
- In vitro/cybrid cells: Sensitization of neuronal cells for degeneration by transfer of dysfunctional mitochondria. In a subclone of the human A549 cell line, devoid of mitochondrial DNA, mitochondria of platelets from PD patients were transplanted. Complex I activity in platelets of PD patients displayed a reduction of 25 % compared with age-matched controls. After transplantation into the A549 cells, complex I activity was reduced by 25% in its activity (Gu et al., 1998).
- In vivo: Induction of mitochondrial dysfunction by Drp1 deletion leads to neuronal cell loss. Maintenance of functional mitochondria in a cell is regulated by fission/fusion processes that allow the elimination of damaged mitochondria and the spreading of intact mitochondria. Deletion of the central fission protein dynamin related protein 1 (Drp1) leads to an elimination in DA neuron terminals in the caudate putamen and to a loss of DA neuron cell bodies in the midbrain. In Drp1 deficient mice, mitochondrial mass decreases, particularly in axons (Berthet et al., 2014) - In vivo: Induction of mitochondrial dysfunction by Tfam knockdown leads to neuronal cell loss. Mitochondrial transcription factor A (Tfam) is a key regulator of mitochondrial biogenesis. Conditional knockout mice with a selective disruption of the gene for mitochondrial Tfam in DA neurons indicated a reduction in mtDNA levels and deficiencies in the respiratory chain in midbrain DA neurons that progressed to DA cell death. The demise of DA neurons in the SNpc was associated with the onset of PD symptoms such as a reduction in locomotor activity of these mice by ca. 30 %. The decrease in locomotor activity was reversed by L-DOPA treatment (Ekstrand et al., 2007).
- In vivo: MPTP dependent mitochondrial dysfunction and cell death is protected by PGC-1 overexpression. Peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) is a key regulator of mitochondrial biogenesis and metabolism. Transgenic mice overexpressing PGC-1 show protection against MPTP intoxication (50 %). The SNpc in these mice is characterized by elevated levels of SOD2, Trx2. Resveratrol is a known activator of SIRT1, leading to enhanced PGC-1 gene transcription. In MPTP mice, resveratrol protected TH-positve neurons by 80% from cell loss (Mudo et al., 2012).
- In vivo: Prevention of mitochondrial dysfunction protects from nigrostriatal cell loss. In order to demonstrate the causative connection between complex I-dependent mitochondrial dysfunction and the degeneration of DA neurons, a series of in vivo experiments were conducted that indicated partial restoration by antioxidants or by compounds supporting a dysfunctional mitochondrial ATP generation. In MPTP challenged mice that additionally received Q10 treatment, a 37 % higher striatal DA level compared with the MPTP group was detected. TH positive staining in the striatum dropped by ca. 65 % after MPTP. In the MPTP + Q10 group, the loss in striatal TH staining was reduced to ca. 40 % compared with the untreated controls. (Beal et al., 1998). In MPTP challenged marmosets, TH positive cell body numbers were reduced by ca. 60 %, co-administration with ebselen resulted in a reduction of TH staining of only ca. 25 % (Moussaoui et al., 2000). In MPTP challenged mice, a reduction of striatal DA by ca. 70 % was detected. Co-treatment with creatine resulted in a reduction of DA levels of only 42 %. In the same setup, TH positive neuron number in the SNpc was reduced by 70 % in response to MPTP, in the presence of creatine, a drop of only 4 % was observed (Matthews et al., 1999).
- In vivo/rotenone: Antioxidants prevent from rotenone-dependent nigrostriatal cell death. Rotenone administered subcutaneously for 5 weeks (2.5 mg/kg/d) caused a selective increase in oxidative damage in the striatum as compared to the hippocampus and cortex, accompanied by massive degeneration of dopaminergic neurons in the substantia nigra. Antioxidant polydatin (Piceid) treatment significantly prevented the rotenone-induced changes in the levels of glutathione, thioredoxin, ATP, malondialdehyde and the manganese superoxide dismutase (SOD) in the striatum, confirming that rotenone- induced mitochondrial dysfunction resulted in oxidative stress (Chen et al., 2015).
- In vivo/rotenone: Degeneration of DA neurons depends on oxidative stress evoked by mitochondrial dysfunction. Many studies have shown that mitochondrial aldehyde dehydrogenase 2 (ALDH2) functions as a cellular protector against oxidative stress by detoxification of cytotoxic aldehydes. Dopamine is metabolized by monoamine oxidase to yield 3,4-dihydroxyphenylacetaldehyde (DOPAL) then converts to a less toxic acid product by ALDH. The highly toxic and reactive DOPAL has been hypothesized to contribute to the selective neurodegeneration of dopamine (DA) neurons. In this study, the neuroprotective mechanism of ALDH2 was observed as overexpression of wild-type ALDH2 gene, but not the enzymatically deficient mutant ALDH2*2 (E504K), reduced rotenone-induced DA neuronal cell death. Application of a potent activator of ALDH2, Alda-1, was effective in protecting against rotenone-induced (100 nM, 24 hr exposure) apoptotic cell death in both SH-SY5Y cells and primary cultured substantia nigra (SN) DA neurons. These results were confirmed by in vivo studies. Intraperitoneal administration of Alda-1 to C57BL/6 mice treated with rotenone (50 mg/kg/day, oral administration for 14days) or MPTP (40 mg/kg/day, i.p. for 14 days) significantly reduced death of SN tyrosine hydroxylase-positive dopaminergic neurons. The attenuation of rotenone-induced apoptosis by Alda-1 resulted from decreasing ROS accumulation, reversal of mitochondrial membrane potential depolarization, and inhibition of activation of proteins related to mitochondrial apoptotic pathway. The present study demonstrates that rotenone or MPP+ induces DA neurotoxicity through oxidative stress. Moreover, Alda-1 is effective in ameliorating mitochondrial dysfunction by inhibiting rotenone or MPP+ induced mitochondria-mediated oxidative stress that leads to apoptosis (Chiu et al., 2015).
Human studies: PD patients were found to show striatal oxidative stress directly relating to the progression of disease severity (Ikawa et al., 2011), suggesting that oxidative stress may cause synaptic dysfunction. Indeed, interruption of the activity-driven local ATP synthesis by synaptic mitochondria (an auto-regulated mechanism) can impair synaptic function (Rangaraju et al., 2014).
Additional empirical support added after the external review:
- Humans + MPTP. Neuropathological examination reveals severe depletion of neurons of the SNpc, no Lewy bodies, clustering of glia; indications for self-perpetuating neurodegeneration following initiation by MPTP. Patients display Parkinsonian motor deficits; responsive to L-Dopa (Ann Neurol 1999, 46(4), 588-605). Nigrostriatal DA neurodegeneration confirmed in live patients by PET analysis (Ann Neurol 1994, 36(5), 765-770).
- Mice/rats + MPTP. Intrastriatal infusion of MPP+. EC50 for DA neurotoxicity was 0.4 mM (mice) and 4.3 mM (rats). Depletion of striatal DA reflected this ca. 10-fold difference between mice and rats (JPET 1994, 270(3), 1008-1014).
- Rats/intranasal infusion of MPTP (0.1 mg/nostril). Reduction in DA neurons in the olfactory bulb and the substantia nigra by ca. 50 %. Reduction in DA by ca. 50 %. Correlation of nigra-striatal DA neuron loss with the onset of impaired motor functions (Exp Neurol 2006, 202(2), 391-403).
- Rats + intranasal MPTP. Decrease in TH staining in the olfactory bulb and SNpc by ca. 30 %. Reduction of DA by ca. 50% in olfactory bulb and 25 % in striatum. Correlated with olfactory, cognitive, and motor impairments (Ann NY Acad Sci 2009, 1170, 629-636).
- Mice + MPTP. Strain differences in the response towards MPTP. Comparison of C57/bl vs. CD 1 mice. C57/bl mice displayed 90 % reduction in striatal DA, while CD1 mice exhibited a reduction of only 30 %. TH expression levels reduced by 45 % in C57/bl mice vs. 27 % reduction in CD1 mice (Exp Neurol 1994, 126(2), 195-204).
- Mice + MPTP. Mice lacking DAT are resistant towards MPTP. Loss of TH-positive neurons in the nigrostriatal system by 34 % in wt (+/+) mice; loss of 22.5% in DAT (+/-) mice; loss of 0 % in DAT (-/-) mice (Exp Neurol 1999, 155, 268-279).
- Rats + rotenone. Loss of striatal fibers (54 %), loss of nigral neurons (28.5 %). TH staining intensity reduced by 50 % in the striatum, reduced by 70 % in the substantia nigra (J. Neurochem 2003, 84, 491-502).
- Rats + rotenone (low 1.5 mg/kg/day vs. high 2.5 mg/kg/day) Dose-dependent loss of striatal DA, loss of TH immunoreactivity in the striatum; dose-dependent catalepsy (Behav Brain Res 2002, 136, 317-324).
- C57/bl 6 mice + rotenone. Oral administration of rotenone for 28 days leads to nigra-striatal DA neurodegeneration, correlation with the onset of motor deficits (J. Neurochem 2007, 101, 1491-1504).
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
Data were retrieved from assays measuring endpoints relevant for AOP3 in relevant cell types.
KE177 Mitochondrial dysfunction
Assays
The following assay endpoints were included:
1. Oxygen consumption rate (in intact cells). This was considered the most reliable method to measure a direct effect on mitochondrial respiration.
2. Image-based measurement of mitochondrial membrane potential (MMP). These methods use dyes such as TMRE or rhodamine123.
3. Measurement of ATP levels. This was considered a more indirect measurement of mitochondrial function, as many cell types can generate ATP via glycolysis in glucose medium, which is independent of mitochondrial function.
The following assay endpoints were deemed not suitable for KE177:
• Resazurin: Indirect measure for KE177, the bioreduction of the dye depends on different sources among which mitochondria
• Lactate dehydrogenase release: Indirect measure for KE177, linked to cell viability
• Extracellular acidification rate: acidification/elevation of glycolysis can also be the consequence of an elevated energy demand that can no longer be met by mitochondria. Under these conditions, an elevation of acidification does not correlate with dysfunction of mitochondria.
KE 890 Degeneration of dopaminergic neurons of the nigrostriatal pathway
Assays
The following assay endpoints were included:
-
Image-based measurement of viability, such as with Calcein-AM or propidium iodide.
-
Image-based measurement of neurite integrity
-
Resazurin
-
Lactate dehydrogenase release
KE 177-KE 890 Cell Models
Neuronal cell models were considered, which included the LUHMES and the SH-SY5Y cells. Data from peripheral neurons were not included, because no information was available on KE 1542 and KE 177, hence it cannot be determined whether KE 890 was affected via KE 1542/177 or via other mechanisms.
Stressors identification
Toxicity data were extracted from Delp et al. (2019, 2021), Bennekou- ENV/JM/MONO(2020)23, van der Stel- ENVJMMONO(2020)22, van Der Stel et al. (2020), Tebby et al. (2022). Data were carefully evaluated by subject-experts to determine whether the chemical affects KE1542 (cIII inhibitors), KE887 (cI inhibitors), KE177 and KE890. Five cI and four cIII inhibitors with evidence of interference with KE1542, KE887, KE177 and KE890 were included as stressors in the assessment and subsequently described. An overview of these data across AOPs and KEs, summarising the percentage effect on each KE, is presented in the “Evidence assessment” and "Quantitative understanding" sections of AOP 3 (cI inhibitors) and AOP 587 (cIII inhibitors).
cI inhibitors
Deguelin
LUHMES
KE 177: Multiple studies measured the OCR in differentiating LUHMES immediately after treatment with deguelin. The effective concentrations range from 0,016 µM to 10 µM (Delp 2021, Alimohammadi 2023, Tebby 2022). When cells were treated for 24 h in glucose containing medium, ATP content was affected in 8-10 µM (ENVJMMONO(2020)22, Delp 2021). This value dropped to 0,0045 µM (2000-fold) when cells were cultured in galactose containing medium (ENVJMMONO(2020)22,)
KE 890: in multiple studies, neurite outgrowth (during 24 h of exposure) was affected at 3-6 µM, with viability being affected at 30 µM or higher. When exposed for 120 h, neurites were affected at 0,40 µM and viability at 1,2 µM (Delp 2021). In galactose containing medium, neurites were affected at 0,007-0,010 µM and viability at 0,15-0,31 µM when exposed to deguelin for 24 h.
SH-SY5Y
In differentiated SH-SY5Y cells, 24 h exposure to deguelin lead to a decrease in mitochondrial membrane potential as measured via rhodamine 123 with an EC25 of 0,31 µM, but ATP content was not affected up to 10 µM, which was the highest tested concentration. A prolonged exposure to deguelin (120 h) however, affected the ATP content with an EC50 of 1 µM.
KE4 is not affected in differentiated SH-SY5Y when exposed to deguelin for 24 h. Prolonged exposure (120 h) however disturbed neurites (EC25 = 3,1 µM) and cell viability (EC = 3-10 µM).
|
KE |
cell type |
assay |
exposure |
effect level |
effect conc |
reference |
notes |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
EC25 |
0,016 µM |
Alimohammadi et al. 2023; Fig S4 |
5 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
10 µM |
Delp_2021, Figure 8ABC |
4 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
EC50 |
~ 0,1 µM |
ENVJMMONO(2020)22 van der Stel, Fig6AB / Annex 1.2 Tebby 2022, Fig4 |
1, 5 |
|
KE 177 |
LUHMES DoD3 |
ATP content |
DoD2-DoD3 (24h) |
EC25 |
10 µM |
Delp 2021, Fig S7+Fig4 |
|
|
KE 177 |
LUHMES DoD3 |
ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
7,8 µM |
ENVJMMONO(2020)22 van der Stel, Fig11 + Fig13 |
|
|
KE 177 |
LUHMES DoD3 |
ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0,0045 µM |
ENVJMMONO(2020)22 van der Stel, Fig13 |
|
|
KE 177 |
SH-SY5Y DoD7 |
mitochondrial membrane potential (via rhodamine123) |
DoD6-DoD7 (24h) |
EC25 |
0,316 µM |
Delp_2021 Figure 7 ENVJMMONO(2020)22 van der Stel Fig8 / Annex 1.6 |
2 |
|
KE 177 |
SH-SY5Y DoD7 |
ATP content |
DoD6-DoD7 (24h) |
EC50 |
> 10 µM |
ENVJMMONO(2020)22 van der Stel, Fig10 |
|
|
KE 177 |
SH-SY5Y DoD8 |
ATP content |
DoD3+DoD6-DoD8 (120h) |
EC50 |
1 µM |
ENVJMMONO(2020)22 van der Stel, Fig10 / Annex 1.16 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
3,1-5,6 µM (neurites) 31,6-34,5 µM (viability) |
Delp_2019 Figure 4 ENVJMMONO(2020)22 van der Stel Fig13 |
2 |
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
4,0 µM (neurites) borderline at 50 µM (viability) |
Delp_2021 Figure 2+4 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
~ 3 µM (neurites) |
Tebby 2022, Fig 4 |
1 |
|
KE 890 |
LUHMES DoD10 |
NeuriTox (+Glc) |
DoD5+DoD7-DoD10 (120 h) |
EC25 |
0,40 µM (neurites) 1,26 µM (viability) |
Delp_2021, Figure S3 |
|
|
KE 890 |
LUHMES DoD3 |
Resazurin |
DoD2-DoD3 (24h) |
EC25 |
31 µM |
Delp 2021, Supplement Figure S7 + S3 ENVJMMONO(2020)22 van der Stel, figure 13 |
2 |
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0,01 µM (neurites) 0,31 µM (viability) |
Delp_2019, Figure 4 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0,0074 µM (neurites) 0,15 µM (viability) |
ENVJMMONO(2020)22 van der Stel, Fig13 / Annex 1.14 |
|
|
KE 890 |
SH-SY5Y DoD7 |
NeuriTox |
DoD6-DoD7 (24h) |
EC25 |
> 100 µM (neurites) |
Delp_2021 Figure 3 (+Fig S3C) |
|
|
KE 890 |
SH-SY5Y DoD7 |
Lactate production |
DoD6-DoD7 (24h) |
inconclusive |
ENVJMMONO(2020)22 van der Stel, figure 9 |
||
|
KE 890 |
SH-SY5Y DoD7 |
Resazurin |
DoD6-DoD7 (24h) |
EC25 |
> 10 µM |
Delp_2021 Figure 7 |
|
|
KE 890 |
SH-SY5Y DoD8 |
NeuriTox |
DoD3+DoD6-DoD8 (120h) |
EC25 |
3,1 µM (neurites + viability) |
Delp_2021 Figure 3 (+Fig S3C) |
3 |
|
KE 890 |
SH-SY5Y DoD8 |
NeuriTox |
DoD3+DoD6-DoD8 (120h) |
EC50 |
10 µM (viability via propidium iodide) |
ENVJMMONO(2020)22 van der Stel, Fig10 / Annex 1.16 |
3 |
DoD: Day of Differentiation
1EC estimated visually from graph.
2Likely same underlying data reprinted
3Unclear if same data reprinted
4Only n=2
5Number of biological replicates unclear
Rotenone
LUHMES
KE177: Multiple studies measured the OCR in differentiating LUHMES immediately after treatment with rotenone. The lowest effective concentration was at 10 nM. When cells were treated for 24 h, OCR was affected at 100 – 500 nM. In the same exposure scenario, MMP was 24 nM, but ATP levels only decreased at 5-40 µM, when cells were cultured with glucose-containing medium. When fed with galactose, cells were more sensitive, with MMP and ATP affected at 0.2 nM and 0.01-6 nM, respectively. This corresponds to a shift of 200 to 1000-fold.
KE890: In multiple studies, neurite outgrowth (during 24 h of exposure) was affected at 20-200 nM, with viability being affected at 3-30 µM or higher. When exposed for 120 h, neurites were affected at 31 nM and viability at 250 nM. In galactose containing medium, neurites were affected at 0,3-3 nM and viability at 65-300 nM when exposed to rotenone for 24 h. This corresponds to a shift of about 100-fold.
SH-SY5Y
KE177: In differentiated SH-SY5Y cells, 24 h exposure to rotenone lead to a decrease in mitochondrial membrane potential at 40-100 nM as measured via rhodamine 123, but ATP content was not affected up to 10 µM, which was the highest tested concentration. A prolonged exposure to rotenone (120 h) however, affected the ATP content at 100 nM.
KE890: Neurite integrity was affected at 16 nM, but viability was not affected up to 10 µM (highest tested concentration) in the 24 h exposure scenario. Prolonged exposure (120 h) however disturbed neurites at about the same concentration (25 nM) and cell viability in the higher nanomolar range (EC = 40-1000 nM). To summarize, rotenone is potent in disturbing the mitochondrial membrane potential and neurite integrity, but an effect on ATP and viability can only be observed upon prolonged exposure.
|
KE |
cell type |
assay |
exposure |
effect level |
effect conc |
reference |
notes |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
10 µM |
Delp 2021, Figure 8 ABC |
|
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
10 µM |
ENVJMMONO(2020)22 van der Stel, Fig6AB / Annex 1.2 |
2 |
|
KE 177 |
LUHMES (intact) |
OCR |
immediate |
EC50 |
0.01 µM (basal respiration) |
Tebby 2022, Table S1 |
|
|
KE 177 |
LUHMES DoD3 |
OCR |
DoD2-DoD3 (24h) |
EC50 |
0.5 µM (basal respiration) 0.1 µM (maximal respiration) |
ENVJMMONO(2020)22 van der Stel, Fig6 C |
|
|
KE 177 |
LUHMES DoD3 |
MMP (via TMRE) (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
0.024 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
MMP (via TMRE) (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.0002 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
25.12 µM |
Delp 2019, Figure 3 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content |
DoD2-DoD3 (24h) |
EC25 |
5.011 µM |
Delp 2021, Figure 4 + Figure S7 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
40 µM |
ENVJMMONO(2020)22 van der Stel, Figure 13 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
> 1 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0.0056 µM = 5,6 nM |
Delp 2019, Figure 3 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0.0048 µM = 4,8 nM |
ENVJMMONO(2020)22 van der Stel, Figure 13 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.00001 µM = 0,01 nM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
ATP production |
DoD2-DoD3 (24h) |
EC50 |
0.1 µM |
ENVJMMONO(2020)22 van der Stel, Fig6 C |
|
|
KE 177 |
LUHMES DoD3 |
ATP Production |
DoD2-DoD3 (24h) |
EC50 |
100 µM |
ENVJMMONO(2020)22 van der Stel, Fig 11 |
|
|
KE 177 |
SH-SY5Y DoD7 |
MMP (Rhodamine 123) |
DoD6-DoD7 (24h) |
EC25 |
0.040 µM |
Delp 2021, Figure 7 |
|
|
KE 177 |
SH-SY5Y DoD7 |
MMP (Rhodamine 123) |
DoD6-DoD7 (24h) |
EC50 |
0.1 µM |
ENVJMMONO(2020)22 van der Stel, Fig8 Annex 1.6 |
|
|
KE 177 |
SH-SY5Y DoD7 |
intracellular ATP content |
DoD6-DoD7 (24h) |
EC50 |
>10µM |
ENVJMMONO(2020)22 van der Stel, Fig 10 Annex 1.6 |
|
|
KE 177 |
SH-SY5Y DoD8 |
intracellular ATP content |
DoD3-DoD8 (120h) |
EC50 |
0.1µM |
ENVJMMONO(2020)22 van der Stel, Fig 10 Annex 1.6 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+ Glc) |
DoD2-DoD3 (24h) |
EC50 |
31 µM (viability) 0,31 µM (neurites) |
Delp 2019, Figure 3 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+ Glc) |
DoD2-DoD3 (24h) |
EC25 |
>10µM (viability) 0.079 µM (neurites) |
Delp 2021, Figure 2+4 Delp 2021, Figure 2+4 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+ Glc) |
DoD2-DoD3 (24h) |
EC50 |
3.6µM (viability) 0.2µM (neurites) |
ENVJMMONO(2020)22 van der Stel, Fig13 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+ Glc) |
DoD2-DoD3 (24h) |
EC25 |
>1µM (viability) 0.021µM (neurites) |
Alimohammadi 2023, Fig 12 + S1 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox |
DoD2-DoD3 (24h) |
EC50 |
0.3116µM (neurites) |
Tebby 2022, Table S1 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0.3162 µM (viability) 0.003162 µM (neurites) |
Delp 2019, Figure 3 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0.76µM (viability) 0.0025µM (neurites) |
ENVJMMONO(2020)22 van der Stel, Fig13 / Annex 1.14 |
|
|
KE 890 |
LUHMES DoD3 |
NeuriTox (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.065µM (viability) 0.0003µM (neurites) |
Alimohammadi 2023, Fig 12 + S1 |
|
|
KE 890 |
LUHMES DoD3 |
Viability |
DoD2-DoD3 (24h) |
EC50 |
>10µM |
ENVJMMONO(2020)22 van der Stel, Fig 6C |
|
|
KE 890 |
LUHMES DoD3 |
Resazurin |
DoD2-DoD3 (24h) |
EC25 |
1µM |
Delp 2021, Figure S7 |
1 |
|
KE 890 |
LUHMES DoD3 |
Resazurin (+ Glc) |
DoD2-DoD3 (24h) |
EC50 |
10µM (borderline) |
ENVJMMONO(2020)22 van der Stel, Fig13 |
|
|
KE 890 |
LUHMES DoD10 |
NeuriTox |
DoD5-7 + DoD7-10 (120h) |
EC25 |
0.25µM (viability) 0.03162µM (neurites) |
Delp 2021, Figure S3 |
1 |
|
KE 890 |
SH-SY5Y DoD7 |
neurite integrity |
DoD6-DoD7 (24h) |
EC25 |
0.0158 µM (neurites) |
Delp 2021, Figure 3 (+Fig S3C) |
|
|
KE 890 |
SH-SY5Y DoD7 |
Viability |
DoD6-DoD7 (24h) |
EC50 |
>10µM |
ENVJMMONO(2020)22 van der Stel, Fig10 / Annex 1.6 |
|
|
KE 890 |
SH-SY5Y DoD7 |
Resazurin |
DoD6-DoD7 (24h) |
EC25 |
>10µM |
Delp 2021, Figure 7 |
|
|
KE 890 |
SH-SY5Y DoD7 |
LDH, Lactate supernatant, renormalized |
DoD6-DoD7 (24h) |
EC50 |
>10µM |
ENVJMMONO(2020)22 van der Stel, Fig9 / Annex 1.8 |
|
|
KE 890 |
SH-SY5Y DoD8 |
NeuriTox |
DoD3-DoD6 + D6-D8 (120h) |
EC25 |
0.0398 µM (viability) 0.025 µM (neurites) |
Delp 2021, Figure 3 (+Fig S3C) |
|
|
KE 890 |
SH-SY5Y DoD8 |
Viability |
DoD3-6 + DoD6-8 (120h) |
EC50 |
1µM (viability) |
ENVJMMONO(2020)22 van der Stel, Fig10 / Annex 1.16 |
1EC estimated visually from graph.
2Number of biological replicates unclear
Fenpyroximate
LUHMES
Glucose
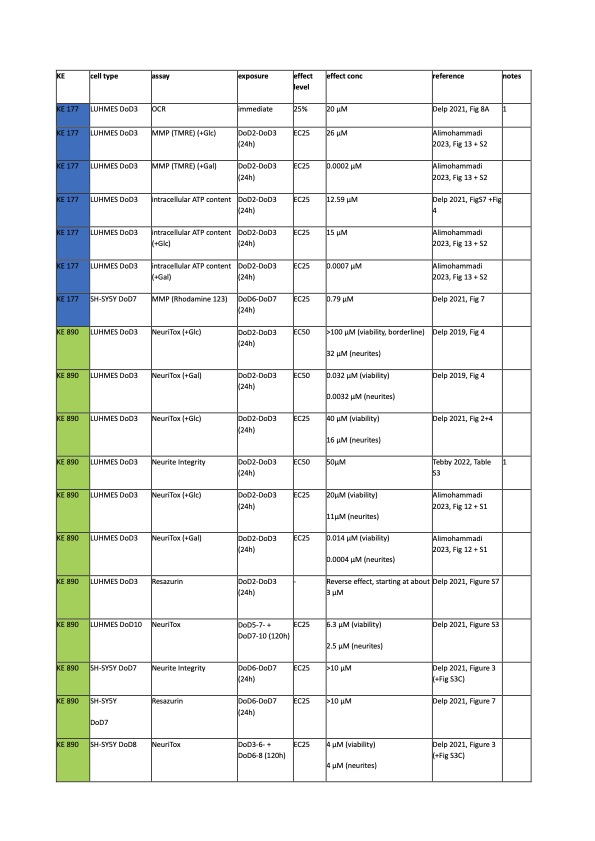
KE177: In intact LUHMES, OCR was reduced with 20 µM fenpyroximate (only 2 biological replicates of a single concentration). MMP, as measured via TMRE, was affected at 26 µM and ATP content at 13-15 µM.
KE890: was measured in multiple studies. Neurite area was reduced at concentrations starting at 11-50 µM. The effect on viability was variable: it ranged from 20 µM to > 100 µM. Prolonged exposure (120 h) to fenpyroximate reduced the EC25 to 2.5 and 6.3 µM, for neurite area and viability, respectively.
Galactose:
KE177: In galactose-containing medium, MMP was affected at 0.2 nM, and ATP was affected at 0.7 nM. These values correspond to a shift of 10.000 fold.
KE890: Neurite area was affected at 0.4 – 3 nM (10.000-fold shift) and viability at 14 – 32 nM (1000-fold shift).
SH-SY5Y
KE177: Only one study investigated the effect of fenpyroximate on KE2. MMP (measured via rhodamine123) was affected at 0.79 µM.
KE890: Neurite integrity and viability was not affected at concentrations up to 10 µM, which was the highest tested concentration. In the prolongued exposure scenario (120 h), viability and neurite integrity were affected both at 4 µM.
Pyrimidifen
LUHMES
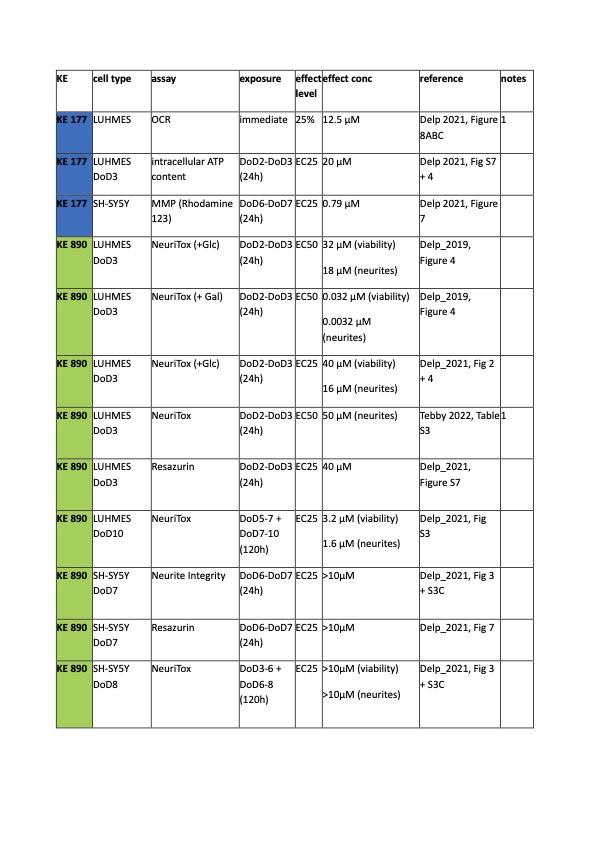
KE177: Only two studies measured KE2 in LUHMES cells: At a concentration of 12.5 uM, pyrimidifen reduced OCR by 75% in intact cells. Of note, this study consisted of only two biological replicates at a single chemical concentration. ATP levels were affected at 20 uM when cells were exposed to pyrimidifen for 24 h.
KE890 was affected in LUHMES cells between 16 – 50 uM when treated from DoD2 to DoD3. In the repeat dosing scenario (120 h), LUHMES were affected at 1.6 uM. LUHMES cultured in galactose medium were sensitive to pyrimidifen at low nanomolar concentrations (0.3 – 3 nM), which corresponds to a shift of 10.000-fold.
SH-SY5Y
Only one study measured KE2 in differentiated SH-SY5Y cells: MMP (measured via rhodamine 123) was affected by pyrimidifen at 0.8 uM.
KE4 was not affected in SH-SY5Y cells, in both the 24 h and the 120 h scenario. In both cases, the highest tested concentration was 10 uM.
Please see Tebufenpyrad and cIII inhibitors in the attached document
- Not endorsed
Uncertainties and Inconsistencies
- Several in vitro studies applying rotenone to evoke mitochondrial dysfunction came to the conclusion that rotenone-dependent ROS formation, and not the rotenone-evoked drop in ATP is the primary cause for cell degeneration. These observations are largely based on experimental systems employing the rotenone insensitive NADH dehydrogenase NDI 1. Expression of NDI 1 protected rotenone exposed cells from degeneration. The presence of NDI 1 however results in a substitution of ATP. Endogenously expressed complex I is still present in these models and it can be assumed that rotenone exposure would still lead to a complex I-dependent formation of ROS that precludes the modeling of a precise cause-consequence relationship between either ATP depletion or elevated ROS levels with the demise of DA neurons.
- Several studies indicate a dominant role of ROS in the degeneration of DA neurons, based on models in which rotenone/MPP+ mediated mitochondrial dysfunction and cell degeneration was protected by the presence of exogenously added antioxidants. Maintenance of the endogenous redox potential however is a highly ATP-dependent process. Clear-cut separations between the respective contribution of ROS or the role of an inhibited mitochondrial ATP synthesis on the degeneration of DA neurons is hence difficult to postulate.
- Studies with chronic partial GSH depletions indicated that an experimental reduction of GSH/GSSG by ca. 50 % has no influence on cell viability. Reports involving rotenone and MPP+ however regularly observe degeneration of DA neurons under conditions of GSH depletion around 50 %. These observations indicate a more prominent role of the intracellular drop of ATP evoked by the complex I inhibitors in the process of cell degeneration.
- Studies in which oxidative stress is generated e.g. by the application of DA or 6-OHDA not only observed a challenge of the cellular redox potential, but also reversible and irreversible inhibitory mechanisms of mitochondrial respiratory chain complexes (nitration, S-nitrosation) that are accompanied by an inhibition of the respiratory chain in the absence of pharmacological complex I inhibitors. These observations illustrate the close mutual interaction between oxidative stress and the inhibition of mitochondrial respiration and point to a profound role of direct mitochondrial inhibition also under oxidative stress conditions.
- Mitochondrial dysfunction is generally associated with conditions of oxidative stress. Dysfunctional mitochondria can act as potent source of superoxide. Oxidative stress associated with PD however not only originates from mitochondrial ROS, but also from DA autoxidation and the Fenton reaction, as well as from inflammatory activated adjacent glia. Interpretations on the role of oxidative stress in DA neurons and its role in DA neurodegeneration is hence hampered by the fact that the respective origin of the reactive oxygen species formed (mitochondria, DA autoxidation, inflammation of glia cells) is rather difficult to identify and often shows overlappings (Murphy et al., 2009; Starkov et al., 2008, Cebrian et al., 2015).
- In PD patients, a reduction in complex I activity in the SNpc, but also in peripheral tissue and cells such as platelets, was reported. Studies with isolated mitochondria indicated that for efficient inhibition of mitochondrial ATP formation, an inhibition of complex I by ca. 70 % is necessary (Davey et al., 1996). Reports on the reduction of complex I activity in PD patients however repeatedly indicated an inhibition of only 25-30 % (Schapira et al., 1989; Schapira et al., 1990; Janetzky et al., 1994).
- Data available on the respective inhibition of the components of the respiratory chain are highly dependent on the experimental setup used. Analysis of mitochondrial respiratory chain complex activities in mitochondrial homogenates provide results different from data obtained with intact, isolated mitochondria. These aspects need to be considered in the interpretation of such data (Mann et al., 1992; Parker et al., 2008; Mizuno et al., 1989; Schapira et al., 1990; Cardellach et al., 1993)
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
|
Uncertainty |
Impact |
Reason |
|
Toxicity data only available from cells cultured in glucose-containing medium |
High |
In vitro cell cultures in general are characterized by an unphysiological high glycolysis rate. In the presence of glucose any KE is influenced by the contribution of oxidative phosphorylation in addition to glycolysis to meet the cellular need for ATP. Thus, the KEs are influenced by the glycolysis rate. Glucose concentrations in culture medium higher than the physiological level enhances cellular resistance to mitochondrial dysfunction. Replacement of glucose with galactose shifts cellular metabolism towards mitochondrial oxidative phosphorylation and increase dependence on mitochondrial respiration. |
|
Time of exposure (24h) |
High |
It is possible that the effects on KE177 occurs more rapidly than that required to observe an effect on KE890. For some of the selected chemicals, the effects start close to the highest tested, concentration after 24 h and continue when exposure is prolonged to 120 h, or may only occur after 120 h. |
|
OCR measured at a single high concentration |
High |
The lack of concentration-response information for KE177 leads to an increased uncertainty in the concordance of the concentration-response relationship across the KEs. |
|
Neurite outgrowth assays (NA) |
Medium |
The assays for KE4 were mostly conducted from DoD2 to DOD3. NA tested on differentiating neurons is not representative of an adult stage. Active molecular mechanisms that are no longer present in adult or differentiated cells are involved in development. A reduction in neurite area may be due to the degeneration of neurites or interference with developing pathways. In this exposure scenario, it is unclear whether the chemical would lead to a loss of neurons, or only a delay in neurite outgrowth. A loss in neurite integrity in the absence of a loss of viability was not considered sufficient to indicate activation of KE 890. |
|
Rotenone |
Medium |
To summarize, the evidence for rotenone affecting the KER in LUHMES is strong, but it should be noted that there is large variation in the determined effective concentrations. For example, ATP production was found to be affected at 100 nM to 100 µM (a range of 1000-fold) in the same study. ATP content in LUHMES cells cultured in galactose medium was affected at 0.01-6 nM (three independent studies), which corresponds to a range of 600-fold. The reasons for these variations in efficacy are currently unknown. |
|
Methodological limits |
Medium-low |
• For some assays and chemicals, only two biological replicates were performed (instead of 3), therefore results should be considered with caution. • In certain studies (i.e., Bennekou 2020), concentration-response data were sometimes “re-normalized”. For many assays, results are normalized to an untreated control, which is set at 100%. But if the results of low concentrations of the solvent chemical are not sufficiently close to 100%, it is assumed that the solvent control was measured imprecise and the curve is re-normalized (i.e., shifted) to reach a 100%. In this situation, some information about the variability in the assay is lost. • Different assays have a different effect concentration (i.e., EC25 and EC50). Occasionally, also the same assay can have different effect levels depending on the publication, which reduces overall comparability. However, in most cases the EC25 and EC50 are within a factor of 3 of each other, thus limiting the uncertainty. |
Additional uncertainties of interest for this document are discussed in Bennekou et al- OECD Series on Testing and Assessment No. 326, Case study on the use of integrated approaches to testing and assessment for identification and characterisation of parkinsonian hazard liability of deguelin by an aop-based testing and read across approach.
- Not endorsed
Known modulating factors
Quantitative Understanding of the Linkage
Quantitative understanding for this KE relationship mainly comes from in-vitro and engineered systems, using rotenone and MPTP as main chemical stressors. A clear response- response effect is evident as well as temporality was mainly supported by evidence that modulation of the KE up was attenuating or preventing the KE down. Evidence of dose relationship was limited, as most of the time a single, generally high, concentration was used.
|
KE 2 upstream |
KE 4 downstream |
Comments |
Reference |
|
Rotenone experiments |
|||
|
Mitochondrial membrane potential reduced by 50 % upon rotenone treatment. Back to 80 % compared to controls in the presence of the flavonoid rutin. Intracellular Ca2+ elevated by a factor of 3 by rotenone, reduction to an increase of 1.5 in the presence of rutin. ROS increased by a factor of 6.5; increase of ROS by a factor of 2 in the presence of rutin. |
Rotenone (10 µM) resulted in a reduction of cell viability by 50 %. In the presence of rutin, cell viability was only reduced by 10 % upon rotenone treatment |
SH-SY5Y cells exposed to rotenone (10 µM) for 24 h. When applied alone, rutin displayed no toxic effects, up to 100 µM. Rutin was added to the cells 30 min prior rotenone at concentrations from 0-10 µM |
Park et al., 2014 |
|
Mitochondrial membrane potential reduced by ca. 66 % upon rotenone treatment; in the presence of celastrol, reduction by ca. 55 %. ROS formation increased by a factor of 2 in the presence of rotenone; ROS increase by a factor of 1.5 in the presence of celastrol. |
Cell viability was reduced by 50 % by rotenone; In the presence of the triterpene celastrol, cell viability was only reduced by ca. 10 % |
SH-SY5Y + rotenone (10 µM). Celastrol (2.5 nM) was applied 90 min prior to rotenone. Cells were incubated with the two compounds for a period of 24 h. |
Choi et al., 2014 |
|
TH staining in the SNpc in arbitrary units: Control (25) Rotenone (14) Rotenone + NDI 1(22) TH staining in the striatum Control (70) Rotenone (40) Rotenone + NDI 1 (65) DA levels in the striatum: Control (2.5) Rotenone (1.3) Rotenone + NDI 1 (2.2) |
5 month old male Sprague-Dawley rats (ca. 500 g) received intracerebral injection of recombinant adeno-associated virus with the NADH dehydrogenase NDI 1 gene. 45 days after virus injection, rats were treated with rotenone-loaded microspheres (poly(DL-lactide-co-glycolide). 100 mg rotenone /kg body weight s.c. With this method, HPLC analysis of plasma rotenone revealed levels of 2 µM 14 days after microsphere treatment, and 1 µM 60 days after microsphere treatment. Behavioral experiments and brain sample collection was conducted 30 days after rotenone treatment. |
Marella et al., 2008 |
|
|
MPP+ experiments |
|||
|
Decline in mitochondrial transmembrane potential by MPP+; 50 % prevention from this decline by rosmarinic acid. NADH levels were reduced by ca. 50 % in the presence of MPP+; loss of NADH was completely prevented by the presence of rosmarinic acid. ROS levels increased by 50 % in the presence of MPP+. Rosmarinic acid lead to a reduced increase of ROS by only 20 % compared with the untreated control. |
Cell viability reduced by MPP+ by 30 %, complete protection by the presence of the antioxidant rosmarinic acid. Striatal DA content reduced by 40 % by MPP+ treatment, partially protected by rosmarinic acid back to a value of 25 % reduction compared with the untreated control. |
MES23.5 cells exposed to MPP+ (200 µM) for 24 h. Rosmarinic acid (1 nM) was applied 30 min prior to MPP+ treatment. |
Du et al. 2010 |
|
Reduction in mitochondrial membrane potential by 60 % (MPP+), by 50 % (rotenone), complete recovery by the co-incubation with ISB, PHT, PHO |
SH-SY5Y + MPP+: Cell viability reduced by 66 %; ISB, PHT, PHO partially protected from cell death with a reduction in cell viability by ca. 20 % SH-SY5Y + rotenone: reduction in cell viability by 60 % Partial protection by ISB, PHT, PHO to a reduction in cell viability by 25-50 %. SH-SY5Y + BSO: Reduction in cell viability by 80 % ISB, PHT, PHO partially protected with a residual decline in cell viability by ca. 20 % |
SH-SY5Y + MPP+ (200 µM) or rotenone (150 nM) or BSO (150 µM) for 60 h and 72 h. Antioxidants tested: Iminostilbene (ISB) Phenothiazine (PHT) Phenoxazine (PHO) The antioxidants were applied 2 h prior to rotenone, MPP+, or BSO treatment |
Hajieva et al., 2009 |
|
Circumvention of endogenous complex I |
|||
|
wt cells exposed to rotenone: increase in carbonyl content as marker of oxidative stress by 100 %; completely prevented in NDI 1 expressing cells. In midbrain slice cultures exposed to rotenone: increase in carbonyl content by 20 % Rats exposed to rotenone: increase in carbonyl content: 27 % in the striatum, increase by 41 % in the midbrain |
SK-N-MC cells: rotenone evoked cell death protected by ca. 90 % in NDI 1 expressing cells. Rotenone induced cell death prevented by 80 % by alpha-tocopherol (62.5 µM and 125 µM). |
SK-N-MC human neuroblastoma cells transfected with the rotenone insensitive NADH dehydrogenase NDI 1; Cells were treated with rotenone (100 nM) for 48 h or with BSO (10 µM) for 24 h. When both compound were used in a combined experiment, cells were first treated with BSO (10 µM) for 24 h, then rotenone (10 nM) was added for additional 36 h. |
Sherer et al., 2003 |
|
Application of the complex I inhibitors: Rotenone Fenazaquin Fenpyroximate Pyridaben Tebufenpyrad Pyridaben |
Time and concentration-dependent cell death with rotenone and a series of other complex I inhibitors. NDI 1 expressing cells were resistant towards the different complex I inhibitors. |
SK-N-MC human neuroblastoma cells expressing the rotenone-insensitive NADH dehydrogenase NDI 1 from saccharomyces cerevisiae. All complex I inhibitors applied were added at the concentrations: 10 nM, 100 nM, 1 µM. Pyridaben was applied at 1 µM, 10 µM, 100 µM. Viability was assessed after 48 h, ATP was detected after 6 h. Carbonyl content was detected after 24 h. |
Sherer et al., 2007 |
|
Oxygen consumption rate doubled by MB in the absence of complex I inhibitor. Oxygen consumption reduced by 50 % by rotenone; completely reversed to control levels by the presence of MB. Complex I-III activity reduced by 95 % by rotenone. Reversed to control levels by the presence of MB. |
HT22 cell viability reduced by 70 % by rotenone. In the presence of MB, reduction by only 10 % of cell viability was observed. In rats treated with rotenone, rotarod retention time was reduced by 50 % by rotenone. Completely reversed to control levels by the co-administration of MB. In rats, rotenone evoked a reduction of striatal DA by 50 %; completely reversed to control levels by MB Complex I-III activity in the striatum of rats was reduced by 50 %, residual inhibition of 10 % observed in rats that were additionally treated with MB |
The study included:
Test of methylene blue (MB) (10 and 100 ng/ml in isolated mitochondria; 1 and 10 µg/ml in HT 22 cells) to circumvent the complex I/III blockade |
Wen et al. 2011 |
|
Cybrid cells with PD mtDNA display a reduction in complex I activity by 20 %. |
Cybrid cells: increase in basal formation of reactive oxygen species by 80%. 2-times higher sensitivity towards MPP+ as stressor |
SH-SY5Y cells devoid of mtDNA; fused with platelets from PD patients for mitochondria transfer: cybrid cells. Treatment with MPP+ (40 or 80 µM) for 24 h or 48 h |
Swedlow et al., 1996 |
|
Oxidative stress causes mitochondrial dysfunction |
|||
|
Isolated mitochondria: Exposure to DA: loss of ca. 50 % membrane potential. Completely protected by GSH or N-acetyl-cystein (NAC) Decline of mitochondrial respiration capacity by 90 %. In the presence of NAC or GSH, only a reduction by 25-30 % was observed. PC12 cells exposed to DA, then isolation and analysis of mitochondria: inhibition of complex I activity by ca. 50 %, prevented by co-incubation with NAC. Inhibition of complex II and III; prevented by NAC. Intact PC12 exposed to DA: Mitochondrial transmembrane potential reduced by ca. 50 %; prevented by NAC Intracellular ATP reduced by ca. 50 %; Cell death increased by DA by ca. 30 %, caspase 3 activity increased by a factor of 3; all increases prevented by the presence of NAC. |
PC12 cells exposed to DA: Increase in intracellular ROS by a factor of 2; completely reversed by NAC Quinoprotein formation increased by a factor of 3; completely prevented by the presence of NAC or GSH. Cell death increased from 3 % (control) to 37 % (DA). Reduced to 10 % in the presence of NAC. |
PC12 cells and isolated rat brain mitochondria exposed to dopamine (100-400 µM). N-acetyl cysteine or GSH for protection were added at a concentration of 2.5 mM. In experiments including isolated mitochondria, NAC and GSH were added 2 h prior to DA. In experiments including PC12 cells, NAC and GSH were added 1 h prior DA. Isolated mitochondria were exposed to DA for 2 h; PC12 cells were expose to DA for 24 h. |
Jana et al., 2011 |
|
Reduction of intracellular GSH by 50 % and of intramitochondrial GSH by 60 % leads to: Mitochondrial ROS increased by 30 % ATP levels reduced by 66 % Mitochondrial activity reduced by 66 % State 3 respiration reduced by 60 % Complex I activity inhibited by 60 % |
Whole cell ROS increased by 30 % |
PC12 cells with inducible knockdown of glutamyl cysteine synthetase (inhibition of GSH synthesis) by addition of 25 µg/ml doxycycline. Treatment for 24 h with doxycycline resulted in a GSH decline by ca. 50 %. |
Jha et al., 2000 |
|
Reduction of GSH levels by ca. 50 % result in: Complex I inhibition by 40 %; completely reversed by DTT. |
No cell toxicity under the applied conditions |
N27 cells exposed to BSO (2.5 µM) for 7 days: Total glutathione was declined by ca. 50 % by this chronic treatment; absence of cell toxicity under these conditions. DTT for restoration of complex I activity was added at 1 mM. |
Chinta et al., 2006 |
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
The quantitative understanding of the KERs was gained by modelling the KERs within the qAOP framework and methods that were developed in Tebby et al., (2022). Data and uncertainties are reported in "quantitative understanding" sections of AOP 3 (cI inhibitor) and AOP 587 (cIII inhibitors).
- Not endorsed
Response-response Relationship
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
An overview of these data across AOPs and KEs, summarising the percentage effect on each KE, is presented in the “Evidence assessment” sections of AOP 3 (cI inhibitor) and AOP 587 (cIII inhibitors).
- Not endorsed
Time-scale
Known Feedforward/Feedback loops influencing this KER
Domain of Applicability
There are no sex or age restiction for the applicability of this KEr and mitochondrial are essential for most of eukariotyc cells. Rotenone and MPTp have been tested successfully in primates and mice. The mouse C57BL/6 strain is the most frequently used strain in the reported experiments. A difference in vulnerability was observed, particularly for rats, depending on the strain and route of administration. The Lewis strain gives more consistency in terms of sensitivity when compared to the Sprague Dawley. In addition to rodents, the pesticide rotenone has been also studied in Caenorhabditis elegans (C.elegans), Drosophila, zebrafish and Lymnaea Stagnalis (L.stagnalis) (Johnson et al., 2015), indicating that the system is preseved across species.
References
Alam M, Schmidt WJ.Behav Brain Res. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. 2002 Oct 17;136(1):317-24.
Alimohammadi Mahshid, Birthe Meyburg, Anna-Katharina Ückert, Anna-Katharina Holzer, Marcel Leist, 2023. EFSA Pilot Project on New Approach Methodologies (NAMs) for Tebufenpyrad Risk Assessment. Part 2. Hazard characterisation and identification of the Reference Point. EFSA supporting publication 2023:EN-7794. 56 pp. doi:10.2903/sp.efsa.2023.EN-7794
Andén NE, Hfuxe K, Hamberger B, Hökfelt T (1966) A quantitative study on the nigro-neostriatal dopamine neuron system in the rat. Acta Physiol Scand. 67(3):306-12.
Antunes F, Han D, Rettori D, Cadenas E. (2002) Mitochondrial damage by nitric oxide is potentiated by dopamine in PC12 cells. Biochim Biophys Acta. 1556(2-3):233-8.
Beal MF, Matthews RT, Tieleman A, Shults CW (1998) Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 783(1):109-14.
Bennekou, S. H., van der Stel, W., Carta, G., Eakins, J., Delp, J., Forsby, A., Kamp, H., Gardner, I., Zdradil, B., Pastor, M., Gomes, J. C., White, A., Steger-Hartmann, T., Danen, E. H. J., Leist, M., Walker, P., Jennings, P., & van de Water, B. (2020).ENV/JM/MONO(2020)23 Case study on the use of integrated approaches to testing and assessment for mitochondrial complex-iii-mediated neurotoxicity of azoxystrobin - read-across to other strobilurins: Series on testing and assessment no. 327. Organisation for Economic Co-operation and Development.
Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K. (2014) Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci. 34(43):14304-17.
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 3(12):1301-6.
Bevan MD, Booth PA, Eaton SA, Bolam JP (1998) Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J Neurosci. 18(22):9438-52.
Bezard E, Gross CE, Fournier MC, Dovero S, Bloch B, Jaber M Exp Neurol. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter.1999 Feb;155(2):268-73.
Bolam JP, Pissadaki EK (2012) Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 27(12):1478-83. Cardellach F, Martí MJ, Fernández-Solá J, Marín C, Hoek JB, Tolosa E, Urbano-Márquez A. (1993) Mitochondrial respiratory chain activity in skeletal muscle from patients with Parkinson's disease. Neurology. 43(11):2258-62.
Bywood PT, Johnson SM. Mitochondrial complex inhibitors preferentially damage substantia nigra dopamine neurons in rat brain slices. Exp Neurol. 2003 Jan;179(1):47-59. doi: 10.1006/exnr.2002.8044. PMID: 12504867.
Caboni P, Sherer TB, Zhang N, Taylor G, Na HM, Greenamyre JT, Casida JE. Rotenone, deguelin, their metabolites, and the rat model of Parkinson's disease. Chem Res Toxicol. 2004 Nov;17(11):1540-8. doi: 10.1021/tx049867r. PMID: 15540952.
Cebrián C, Loike JD, Sulzer D. (2015) Neuroinflammation in Parkinson's disease animal models: a cell stress response or a step in neurodegeneration? Curr Top Behav Neurosci. 22:237-70.
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF, Xu JP (2015) Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Mol Neurodegener. 10:4. doi: 10.1186/1750-1326-10-4.
Chinta SJ, Andersen JK (2006) Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radic Biol Med. 41(9):1442-8.
Chiu CC, Yeh TH, Lai SC, Wu-Chou YH, Chen CH, Mochly-Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS (2015) Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp Neurol. 263:244-53.
Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ (2014) Celastrol from 'Thunder God Vine' protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson's disease. Neurochem Res. 39(1):84-96.
Davey GP, Clark JB. (1996) Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. J Neurochem. 66(4):1617-24.
Delp J, Cediel-Ulloa A, Suciu I, Kranaster P, van Vugt-Lussenburg BM, Munic Kos V, van der Stel W, Carta G, Bennekou SH, Jennings P, van de Water B, Forsby A, Leist M. Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors. Arch Toxicol. 2021 Feb;95(2):591-615. doi: 10.1007/s00204-020-02970-5. Epub 2021 Jan 29. PMID: 33512557; PMCID: PMC7870626.
Delp J, Funke M, Rudolf F, Cediel A, Bennekou SH, van der Stel W, Carta G, Jennings P, Toma C, Gardner I, van de Water B, Forsby A, Leist M. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch Toxicol. 2019 Jun;93(6):1585-1608. doi: 10.1007/s00204-019-02473-y. Epub 2019 Jun 12. PMID: 31190196.
Du T, Li L, Song N, Xie J, Jiang H (2010) Rosmarinic acid antagonized 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in MES23.5 dopaminergic cells. Int J Toxicol. 29(6):625-33.
EFSA Panel on Plant Protection Products and their residues (PPR); Ockleford C, Adriaanse P, Berny P, Brock T, Duquesne S, Grilli S, Hernandez-Jerez AF, Bennekou SH, Klein M, Kuhl T, Laskowski R, Machera K, Pelkonen O, Pieper S, Smith R, Stemmer M, Sundh I, Teodorovic I, Tiktak A, Topping CJ, Wolterink G, Angeli K, Fritsche E, Hernandez-Jerez AF, Leist M, Mantovani A, Menendez P, Pelkonen O, Price A, Viviani B, Chiusolo A, Ruffo F, Terron A, Bennekou SH. Investigation into experimental toxicological properties of plant protection products having a potential link to Parkinson's disease and childhood leukaemia. EFSA J. 2017 Mar 16;15(3):e04691. doi: 10.2903/j.efsa.2017.4691. PMID: 32625422; PMCID: PMC7233269.
Ekstrand M, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. (2007) Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 104(4):1325-30.
Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Molecular Cell. 2009;33:627–638.
Giovanni A, P K Sonsalla and R E Heikkila. Studies on species sensitivity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Part 2: Central administration of 1-methyl-4-phenylpyridinium.Journal of Pharmacology and Experimental Therapeutics September 1994, 270 (3) 1008-1014;
Gu M, Cooper JM, Taanman JW, Schapira AH (1998) Mitochondrial DNA transmission of the mitochondrial defect in Parkinson's disease. Ann Neurol. 44(2):177-86.
Hajieva P, Mocko JB, Moosmann B, Behl C (2009) Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. J Neurochem. 110(1):118-32.
Höglinger GU, Féger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC (2003) Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem. 84(3):491-502.
Inden M, Kitamura Y, Takeuchi H, Yanagida T, Takata K, Kobayashi Y, Taniguchi T, Yoshimoto K, Kaneko M, Okuma Y, Taira T, Ariga H, Shimohama S. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J Neurochem. 2007 Jun;101(6):1491-1504.
Jain A, Mårtensson J, Stole E, Auld PA, Meister A (1991) Glutathione deficiency leads to mitochondrial damage in brain. Proc Natl Acad Sci U S A. 88(5):1913-7.
Jana S, Sinha M, Chanda D, Roy T, Banerjee K, Munshi S, Patro BS, Chakrabarti S (2011) Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson's disease. Biochim Biophys Acta. 1812(6):663-73.
Janetzky B, Hauck S, Youdim MB, Riederer P, Jellinger K, Pantucek F, Zöchling R, Boissl KW, Reichmann H. (1994) Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson's disease. Neurosci Lett. 169(1-2):126-8.
Jha N, Jurma O, Lalli G, Liu Y, Pettus EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK (2000) Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. J Biol Chem. 275(34):26096-101. Kawaguchi Y, Wilson CJ, Emson PC (1990) Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci. 10(10):3421-38.
Johnson ME, Bobrovskaya L. 2015. An update on the rotenone models of parkinson’s disease: Their ability to reproduce features of clinical disease and model gene-environment interactions. 946). 101-16.
Jorge Regueiro, Nair Olguín, Jesús Simal-Gándara, Cristina Suñol. Toxicity evaluation of new agricultural fungicides in primary cultured cortical neurons, Environmental Research, Volume 140, 2015, 37-44, https://doi.org/10.1016/j.envres.2015.03.013.
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr (2006) Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 26(19):5256-64.
Khan FH, Sen T, Maiti AK, Jana S, Chatterjee U, Chakrabarti S (2005) Inhibition of rat brain mitochondrial electron transport chain activity by dopamine oxidation products during extended in vitro incubation: implications for Parkinson's disease. Biochim Biophys Acta. 1741(1-2):65-74.
Khan MM, Raza SS, Javed H, Ahmad A, Khan A, Islam F, Safhi MM, Islam F (2012) Rutin protects dopaminergic neurons from oxidative stress in an animal model of Parkinson's disease. Neurotox Res. 22(1):1-15.
Kita H, Kitai ST (1994) The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res. 636(2):308-19.
Koopman W, Willems P (2012) Monogenic mitochondrial disorders. New Engl J Med. 22;366(12):1132-41. doi: 10.1056/NEJMra1012478. Langston JW, Ballard PA Jr (1983) Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med. 309(5):310.
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD (1992) Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. 115 ( Pt 2):333-42.
Marella M, Seo BB, Nakamaru-Ogiso E, Greenamyre JT, Matsuno-Yagi A, Yagi T (2008) Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. PLoS One. 3(1):e1433.
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T (2009) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 29(2):444-53.
Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF (1999) Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 157(1):142-9.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. (1989) Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Commun. 163(3):1450-5.
Moosmann B, Behl C (2002) Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs. 11(10):1407-35. Moussaoui S, Obinu MC, Daniel N, Reibaud M, Blanchard V, Imperato A (2000) The antioxidant ebselen prevents neurotoxicity and clinical symptoms in a primate model of Parkinson's disease. Exp Neurol. 166(2):235-45.
Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D. (2012) Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci. 69(7):1153-65.
Murphy MP. (2009) How mitochondria produce reactive oxygen species. Biochem J. 417(1):1-13.
Muthane U, Ramsay KA, Jiang H, Jackson-Lewis V, Donaldson D, Fernando S, Ferreira M, Przedborski S.Differences in nigral neuron number and sensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57/bl and CD-1 mice. Exp Neurol. 1994 Apr;126(2):195-204.
Orimo S, Uchihara T, Kanazawa T, Itoh Y, Wakabayashi K, Kakita A, Takahashi H (2011) Unmyelinated axons are more vulnerable to degeneration than myelinated axons of the cardiac nerve in Parkinson's disease. Neuropathol Appl Neurobiol. 37(7):791-802.
Park SE, Sapkota K, Choi JH, Kim MK, Kim YH, Kim KM, Kim KJ, Oh HN, Kim SJ, Kim S (2014) Rutin from Dendropanax morbifera Leveille protects human dopaminergic cells against rotenone induced cell injury through inhibiting JNK and p38 MAPK signaling. Neurochem Res. 39(4):707-18.
Parker WD Jr, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 26(6):719-23.
Parker WD Jr, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. 1189:215-8. Perry TL, Godin DV, Hansen S (1982) Parkinson's disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982 Dec 13;33(3):305-10.
Prediger RD, Rial D, Medeiros R, Figueiredo CP, Doty RL, Takahashi RN. Risk is in the air: an intranasal MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) rat model of Parkinson's disease. Ann N Y Acad Sci. 2009 Jul;1170:629-36. doi: 10.1111/j.1749-6632.2009.03885.x.
Prediger RD, Aguiar AS Jr, Matheus FC, Walz R, Antoury L, Raisman-Vozari R, Doty RL.Intranasal administration of neurotoxicants in animals: support for the olfactory vector hypothesis of Parkinson's disease. Neurotox Res. 2012 Jan;21(1):90-116. doi: 10.1007/s12640-011-9281-8. Epub 2011 Oct 15. Review
Rangaraju V, Calloway N, and Ryan TA. 2014. Activity-driven local ATP synthesis is required for synaptic function. Cell. 156(4)825–835.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 54(3):823-7.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1(8649):1269.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT (2003) Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 23(34):10756-64.
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT (2007) Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J Neurochem. 100(6):1469-79.
Snow BJ, Vingerhoets FJ, Langston JW, Tetrud JW, Sossi V, Calne DB. Pattern of dopaminergic loss in the striatum of humans with MPTP induced parkinsonism. J Neurol Neurosurg Psychiatry. 2000 Mar;68(3):313-6.
Starkov AA (2008) The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 1147:37-52.
Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP Jr, Davis RE, Parker WD Jr (1996) Origin and functional consequences of the complex I defect in Parkinson's disease. Ann Neurol. 40(4):663-71.
Surmeier DJ1, Guzman JN, Sanchez-Padilla J, Goldberg JA. 2010. What causes the death of dopaminergic neurons in Parkinson's disease? Prog Brain Res. 2010;183:59-77. doi: 10.1016/S0079-6123(10)83004-3.
Sulzer D, Surmeier DJ. 2013. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Movement Disorders. 28 (6) 715-24.
Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632.
Tebby C, Gao W, Delp J, Carta G, van der Stel W, Leist M, Jennings P, van de Water B, Bois FY. A quantitative AOP of mitochondrial toxicity based on data from three cell lines. Toxicol In Vitro. 2022 Jun;81:105345. doi: 10.1016/j.tiv.2022.105345. Epub 2022 Mar 10. PMID:35278637.
Tepper JM, Bolam JP (2004) Functional diversity and specificity of neostriatal interneurons. Curr Opin Neurobiol. 14(6):685-92.
Van der Stel W, Carta G, Eakins J, Darici S, Delp J, Forsby A, Bennekou SH, Gardner I, Leist M, Danen EHJ, Walker P, van de Water B, Jennings P. Correction to: Multiparametric assessment of mitochondrial respiratory inhibition in HepG2 and RPTEC/TERT1 cells using a panel of mitochondrial targeting agrochemicals. Arch Toxicol. 2020 Aug;94(8):2731-2732. doi: 10.1007/s00204-020-02849-5. Erratum for: Arch Toxicol. 2020 Aug;94(8):2707-2729. doi: 10.1007/s00204-020-02792-5. PMID: 32720191; PMCID: PMC7645484.
van der Stel, Wanda; Bennekou, Susanne Hougaard; Carta, Giada; Eakins, Julie; Delp, Johannes; Forsby, Anna; Kamp, Hennicke; Gardner, Ian; Zdradil, Barbara; Pastor, Manual (2020) ENV/JM/MONO(2020)22 CASE STUDY ON THE USE OF INTEGRATED APPROACHES TO TESTING AND ASSESSMENT FOR IDENTIFICATION AND CHARACTERISATION OF PARKINSONIAN HAZARD LIABILITY OF DEGUELIN BY AN AOP-BASED TESTING AND READ ACROSS APPROACH Series on Testing and Assessment No. 326
van der Stel W, Yang H, Vrijenhoek NG, Schimming JP, Callegaro G, Carta G, Darici S, Delp J, Forsby A, White A, le Dévédec S, Leist M, Jennings P, Beltman JB, van de Water B, Danen EHJ. Mapping the cellular response to electron transport chain inhibitors reveals selective signaling networks triggered by mitochondrial perturbation. Arch Toxicol. 2022 Jan;96(1):259-285. doi: 10.1007/s00204-021-03160-7. Epub 2021 Oct 13. PMID: 34642769; PMCID: PMC8748354.
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH (2011) Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 286(18):16504-15.
Wu Y, Richard S, Parent A (2000) The organization of the striatal output system: a single-cell juxtacellular labeling study in the rat. Neurosci Res. 38(1):49-62.
Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion and stress. Science. 337:1062–1065.
Zhu C, Vourc'h P, Fernagut PO, Fleming SM, Lacan S, Dicarlo CD, Seaman RL, Chesselet MF (2004) Variable effects of chronic subcutaneous administration of rotenone on striatal histology. J Comp Neurol. 478(4):418-26.