
This AOP is licensed under the BY-SA license. This license allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use. If you remix, adapt, or build upon the material, you must license the modified material under identical terms.
AOP: 3
Title
Inhibition of the mitochondrial complex I of nigro-striatal neurons leads to parkinsonian motor deficits
Short name
Graphical Representation
Additional AOP Exploration Options
Click links below to explore AOP 3, Inhibition of the mitochondrial complex I of nigro-striatal neurons leads to parkinsonian motor deficits in tools offered by third parties.

Point of Contact
Contributors
- Andrea Terron
- Clemens Wittwehr
- Anna Price
- Barbara Viviani
- Giacomo Grumelli
Coaches
OECD Information Table
| OECD Project # | OECD Status | Reviewer's Reports | Journal-format Article | OECD iLibrary Published Version |
|---|---|---|---|---|
| 1.33 | WPHA/WNT Endorsed | Scientific Review | iLibrary link |
This AOP was last modified on October 04, 2025 02:11
Revision dates for related pages
| Page | Revision Date/Time |
|---|---|
| Inhibition, NADH-ubiquinone oxidoreductase (complex I) | October 03, 2025 09:44 |
| Binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I) | March 28, 2018 04:51 |
| Increase, Mitochondrial dysfunction | February 11, 2026 07:06 |
| Proteostasis, impaired | October 16, 2025 02:38 |
| Neuroinflammation | July 15, 2022 09:54 |
| Degeneration of dopaminergic neurons of the nigrostriatal pathway | October 01, 2025 07:04 |
| Parkinsonian motor deficits | March 12, 2018 12:44 |
| Increase, Mitochondrial dysfunction leads to Degeneration of dopaminergic neurons of the nigrostriatal pathway | October 03, 2025 17:52 |
| Binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I) leads to Inhibition, NADH-ubiquinone oxidoreductase (complex I) | August 25, 2017 09:35 |
| Inhibition, NADH-ubiquinone oxidoreductase (complex I) leads to Increase, Mitochondrial dysfunction | October 03, 2025 17:14 |
| Increase, Mitochondrial dysfunction leads to Proteostasis, impaired | October 03, 2025 04:49 |
| Proteostasis, impaired leads to Degeneration of dopaminergic neurons of the nigrostriatal pathway | October 03, 2025 04:34 |
| Degeneration of dopaminergic neurons of the nigrostriatal pathway leads to Parkinsonian motor deficits | October 03, 2025 18:38 |
| Degeneration of dopaminergic neurons of the nigrostriatal pathway leads to Neuroinflammation | October 02, 2017 10:30 |
| Neuroinflammation leads to Degeneration of dopaminergic neurons of the nigrostriatal pathway | August 25, 2017 08:54 |
| MPP+ | December 16, 2016 11:22 |
| Rotenone | November 29, 2016 18:42 |
| Deguelin | October 04, 2025 02:09 |
| Pyrimidifen | October 04, 2025 02:11 |
| Fenpyroximate | March 06, 2019 10:45 |
| Tebufenpyrad | October 04, 2025 02:12 |
Abstract
This Adverse outcome Pathway (AOP) describes the linkage between inhibition of complex I (CI) of the mitochondrial respiratory chain and motor deficit as in parkinsonian disorders. Binding of an inhibitor to complex I has been defined as the molecular initiating event (MIE) that triggers mitochondrial dysfunction, impaired proteostasis, which then cause degeneration of dopaminergic (DA) neurons of the nigro-striatal pathway. Neuroinflammation is triggered early in the neurodegenerative process and exacerbates it significantly. These causatively linked cellular key events result in motor deficit symptoms, typical for parkinsonian disorders, including Parkinson's disease (PD), described in this AOP as an Adverse Outcome (AO). Since the release of dopamine in the striatum by DA neurons of the Substantia Nigra pars compacta (SNpc) is essential for motor control, the key events refer to these two brain structures. The weight-of-evidence supporting the relationship between the described key events is based mainly on effects observed after an exposure to the chemicals rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), i.e. two well-known inhibitors of complex I. Data from experiments with these two chemicals reveal a significant concordance in the dose-response relationships between the MIE and AO and within key events (KEs). Also essentiality of the described KEs for this AOP is strong since there is evidence from knock out animal models, engineered cells or replacement therapies that blocking, preventing or attenuating an upstream KE is mitigating the AO. Similarly, there is proved experimental support for the key event relationships (KERs) as multiple studies performed with modulating factors that attenuate (particularly with antioxidants) or augment (e.g. overexpression of viral-mutated α-synuclein) a KE up show that such interference leads to an increase of KE down or the AO. Information from in vitro and in vivo experiments is complemented by human studies in brain tissues from individuals with sporadic Parkinson's disease (Keeney et al., 2006) to support the pathways of toxicity proposed in this AOP.
AOP Development Strategy
Context
Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
Context – Revision of AOP3: Inhibition of cI leading to parkinsonian motor deficits
In 2017, the European Food Safety Authority (EFSA) started a project aimed at establishing a mechanistic AOP to elucidate the causal relationship between mitochondrial complex I (cI) inhibition and neurotoxic adverse outcomes manifesting in loss of dopamine neurons (EFSA 2017). This resulted in the development of AOP:3 (inhibition of cI leading to parkinsonian motor deficits), which subsequently was endorsed by the Organisation for Economic Co-operation and Development (OECD). The initial conceptualization of AOP3 was supported by empirical evidence derived from studies involving rotenone and MPTP/MPP+. Subsequent studies explored whether additional mitochondrial cI inhibitors could trigger similar neurotoxic effects (Table 1). In particular, AOP:3 became a test/pilot case to provide an in vitro point-of-departure (PoD) for an AOP-informed Integrated Approach to Testing and Assessment, concerning a potential risk of Parkinsonian motor deficits after exposure to Tebufenpyrad (Alimohammadi 2022). These open literature publications provide a valid source of data for implementing the biological plausibility, empirical support and quantitative characterisation of the AOP 3. Its implementation is being conducted under a negotiated procedure with EFSA (Reference: NP/EFSA/PREV/2024/02), which is intended to update AOP 3 by incorporating additional evidence into the AOP wiki.
Table 1. Application of AOP 3 in studies with regulatory relevance
|
Study |
Regulatory relevance |
ETC tested |
Reference |
|
Testing of the in vitro battery aligned with AOP3 |
Testing the applicability of several assays to form the basis of a consensus mitochondrial toxicity testing platform |
Inhibition of cI, cII or cIII |
Delp et al. 2019; van der Stel et al. 2020 |
|
Case study on the use of an IATA for identification and characterization of Parkinsonian hazard |
Read across safety assessment of structurally closely related mitochondrial cI inhibitors |
Inhibition of cI |
ENV/JM/MONO(2020)22, van der Stel, W. (2021) |
|
Testing the predictivity of the downstream events |
Testing the inhibitory potency prediction with the aim to understand how far early KE data can and will predict an AO |
Inhibition of cI, cII or cIII |
Delp et al., 2021 |
|
Calibrate a qAOP to predict downstream KEs |
cI inhibitors |
Tebby et al., 2022 |
|
|
EFSA Pilot Project on New Approach Methodologies |
Margins of Internal Exposure Application to Estimated Brain Exposure Compared to In Vitro PoD |
cI inhibitor |
Alimohammadi et al. 2023 |
|
Hazard assessment |
Identification of the signalling network triggered by mitochondrial perturbation induced by the inhibition of cI, cII or cIII in HepG2 cells |
Inhibition of cI, cII or cIII |
Van der Stel et al., 2022 |
PoD: Poin t of Departure; IATA: Integrated Approaches to Testing and Assessment; cI: complex, I; cII: complex II; cIII: complex III
Not endorsed
Strategy
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02): The starting conceptual model for this project is based on the key scientific sources reported in table 1. These publications provided the initial evidence for this project, which was further expanded through a structured literature review aimed at updating the link between mitochondrial toxicity and parkinsonian motor deficits across AOP 3. The updates have been documented in a dedicated section, in agreement with the AOP3 point of contact.
For well-established MIEs and KEs, evidence was retrieved from seminal publications recommended by domain experts and supplemented by expert knowledge. Additional literature was identified, through a structured, non-systematic search using a stressor-based search strategy to retrieve essentiality data and data linking KE887/KE177 to 890/AO through the selected stressors in in vivo studies. Tailored search strings, detailed in a dedicated section at the end of this document, were designed by two information specialists in collaboration with the project team. For each selected stressor, the information specialists conducted a literature search using a quasi-systematic approach. They employed both textwords and database-specific subject headings where available, across the following databases: PubMed, Embase via Elsevier, Web of Science via Clarivate, and Scopus.
The following criteria were applied to select relevant studies.
Publication type
|
Time |
IN |
Inception – present or 2017- present |
|
Language |
IN |
English |
|
Publication type |
IN |
·Primary research studies ·Reviews |
|
|
OUT |
·Expert opinions ·editorials ·letters to the editor ·conference proceedings and posters ·retracted articles ·PhD thesis |
In vitro studies
|
Study design |
IN |
Any In vitro study design |
|
Population |
IN |
·Only cells of the nervous system (i.e., neuronal population and glial cells) at a mature stage ·All species |
|
OUT |
All except those included |
|
|
Exposure |
IN |
·Identified stressors (Objective 2) ·The exposure must occur during the mature stage |
|
OUT |
·Chemical mixture ·Less than one control and three concentrations tested |
|
|
Endpoints |
IN |
·ETC inhibition ·Mitochondrial dysfunction (i.e., oxygen consumption rate, mitochondrial membrane potential, elevated reactive oxygen species, mitochondrial oxidative damage) ·Degeneration of dopaminergic neurons |
In vivo studies
|
Study design |
IN |
Any |
|
OUT |
None |
|
|
Population |
IN |
Mammals and zebrafish |
|
OUT |
All except those included |
|
|
Exposure |
IN |
·Identified stressors. ·The exposure must occur in adults. |
|
OUT |
·In-uterus, developmental stage. ·Mixtures. ·Less than one control and two concentrations tested |
|
|
Endpoints |
IN |
·Labeling of dopaminergic neurons by fluorescent dopamine analogs, or genetically labeled dopaminergic neurons (e.g., GFP expression under control of TH promoter). ·Degeneration of dopaminergic neurons ·Motor deficits |
To develop the empirical evidence, chemicals listed in Delp 2019 and Delp 2021 were considered. In addition, for compounds that were identified or measured as cI inhibitor, data were extracted from Alimohammadi et al. (2023); van der Stel-OECD (2020), van der Stel, W. (2021), Van der Stel et al., (2022) and Tebby et al. (2022). Endpoints and assays were selected on their relevance to AOP 3 and the use of appropriate cell models (i.e., neuronal cells and HepG2 for KE1, neuronal cells only for KE2 and KE4). Further details are provided in the KE section “How it is measured” and in the empirical evidence for the KER.
Quantitative understanding of the KERs was gained by modelling the KERs within the qAOP framework and methods that were developed in Tebby et al. (2022) and further developed during the negotiated procedure with EFSA (Reference: NP/EFSA/PREV/2024/02). A set of compounds used for AOP quantification was selected based on availability of multiple-concentration data representing at least two identical adjacent KEs. Equations representing the KERs were selected based on the dose-response data for adjacent KEs. These equations were parameterized using a bayesian framework, which allowed completing data gaps for cIII inhibition with prior knowledge on cI inhibition.
- Not endorsed
Summary of the AOP
Events:
Molecular Initiating Events (MIE)
Key Events (KE)
Adverse Outcomes (AO)
| Type | Event ID | Title | Short name |
|---|
| MIE | 888 | Binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I) | Binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I) |
| KE | 887 | Inhibition, NADH-ubiquinone oxidoreductase (complex I) | Inhibition, NADH-ubiquinone oxidoreductase (complex I) |
| KE | 177 | Increase, Mitochondrial dysfunction | Increase, Mitochondrial dysfunction |
| KE | 889 | Proteostasis, impaired | Proteostasis, impaired |
| KE | 188 | Neuroinflammation | Neuroinflammation |
| KE | 890 | Degeneration of dopaminergic neurons of the nigrostriatal pathway | Degeneration of dopaminergic neurons of the nigrostriatal pathway |
| AO | 896 | Parkinsonian motor deficits | Parkinsonian motor deficits |
Relationships Between Two Key Events (Including MIEs and AOs)
| Title | Adjacency | Evidence | Quantitative Understanding |
|---|
Network View
Prototypical Stressors
Life Stage Applicability
| Life stage | Evidence |
|---|---|
| Adult | High |
Taxonomic Applicability
Sex Applicability
| Sex | Evidence |
|---|---|
| Mixed | High |
Overall Assessment of the AOP
Domain of Applicability
This proposed AOP is neither sex-dependent nor associated with certain life stage; however, aged animals may be more sensitive. The relevance of this AOP during the developmental period has not been investigated. In vivo testing has no species restriction. The mouse was the species most commonly used in the experimental models conducted with the chemical stressors; though experimental studies using alternative species have been also performed. (Johnson et al. 2015). However, animal models (rodents in particular) would have limitations as they are poorly representative of the long human life-time as well as of the human long-time exposure to the potential toxicants. Human cell-based models would likely have better predictivity for humans than animal cell models. In this case, toxicokinetics information from in-vivo studies would be essential to test the respective concentrations in-vitro on human cells.
Essentiality of the Key Events
Essentiality of KEs for this AOP is strong. There is ample evidence from knock out animal models, engineered cells or replacement therapies that blocking, preventing or attenuating an upstream KE is mitigating the AO. In addition, there is experimental support for the KERs as multiple studies performed with modulating factors that attenuate (particularly with antioxidants) or augment (e.g. overexpression of viral-mutated α-synuclein) a KE show that such interference leads to an increase of KE down or the AO.
|
2 Support for Essentiality of KEs |
Defining Question Are downstream KEs and/or the AO prevented if an upstream KE is blocked ? |
High (Strong) |
Moderate |
Low(Weak) |
|
Direct evidence from specifically designed experimental studies illustrating essentiality for at least one of the important KEs (e.g. stop/reversibility studies, antagonism, knock out models, etc.) |
Indirect evidence that sufficient modification of an expected modulating factor attenuates or augments a KE leading to increase in KE down or AO |
No or contradictory experimental evidence of the essentiality of any of the KEs |
||
|
KE1 Inhibition of complex I |
STRONG |
Rationale: Inactivation of the NADH:Ubiquinone Oxidoreductase Core Subunit S7 (Ndufs 4 gene knockout mice) that produces CI deficiency causes encephalomyopathy, including ataxia and loss of motor skills (Kruse et al., 2008). NDI1-transducted SK-N-MC cells expressing the rotenone-insensitive single subunit NADH dehydrogenase of yeast (NDI1) that acts as a replacement for the entire CI in mammalian cells were completely resistant to 100 nM rotenone, 100 nM fenpyroximate or 1 uM tebufenpyrad-mediated cell death (at 48 hrs of exposure) indicating that cI inhibitors – induced toxicity requires their biding of CI (Sherer et al., 2003). In all rotenone models, mitochondria CI is inhibited at the dose that cause neurodegeneration (Betarbet et al 2000 and 2006). - Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):The mouse model, MCI-Park, by selectively deleting Ndufs2, an essential subunit of MCI, in dopaminergic neurons using intersectional genomics, reproduces several key features of progressive parkinsonism, including impaired dopamine release from striatal axons and deficits in associative learning. These mice initially displayed only mild fine motor deficits, while severe movements impairment responsive to levo-dopa emerged later in life, mirroring aspects of PD pathology. This progression coincided with the spread of dopaminergic signaling deficits from the striatum to the substantia nigra (Gonzalez-Rodriguez et al. 2020). - Not endorsed |
||
|
KE2 Mitochondrial dysfunction |
STRONG |
Rationale: Many studies showing that antioxidants protect the cells against cI inhibitors induced oxidative stress are published (Chen et al. 2015; Lu et al., 2015; Saravanan et al., 2006; Chiu et al., 2015, Sherer et al.2003, Nataraj et al.2015, Wu et al. 1994; Tseng et al. 2014; Li et al. 2010; Kim-Han et al. 2011). This provides (indirect) evidence for essentiality of KE2, if production of reactive oxygen species (ROS) is assumed as direct consequence/sign of mitochondrial dysfunction. Additional evidence comes from experiments with overexpression or activation of antioxidative enzymes (e.g.SOD or ALDH2) , which also prevent rotenone and MPTP/MPP+ induced neurotoxicity (Mudo et al. 2012; Ciu CC et al. 2015). Furthermore, promotion of mitochondrial fusion or blocking of mitochondrial fission prevents or attenuates rotenone and MPTP/MPP+ induced neurotoxicity (Tieu K. et al. 2014). |
||
|
KE3 Impaired proteostasis |
MODERATE |
Rationale: Indirect evidence for the role of disturbed alpha-synuclein proteostasis: Lacking of alpha-synuclein expression in mice prevented induction of behavioural symptoms, neuronal degeneration in the nigrostriatal pathway and loss of DA neurons after chronic treatment with MPTP/MPP+ (Fornai et al. 2004; Dauer et al. 2002) . Injection of adeno/lenti-associated virus that expresses wild-type or mutant α-synuclyn into rat, mice or non-human primate SN produced loss of dopaminergic neurons, but the effect is not easily reproduced in transgenic mice overexpressing alpha-synuclein (Kirk, 2002; Klein, 2002; Lo Bianco, 2002; Lauwers, 2003; Kirk, 2003). Rationale for the role of autophagy: Early dendritic and axonal dystrophy, reduction of striatal dopamine content, and the formation of somatic and dendritic ubiquitinated inclusions in DA neurons were prevented by ablation of Atg7 (an essential autophagy related gene (Friedman et al. 2012)). Rationale for the role of Ubiquitin Proteosomal System/Autophagic Lysosomal Pathway (UPS/ALP): Protection from DA neuronal death was also observed in multiple experiments through the pharmacological modulation of the UPS, ALP system; however, there are also contradicting data in the literature. (Inden et al. 2007; Fornai et al. 2003; Dehay et al. 2010; Zhu et al. 2007, Fornai et al. 2005). However, although many lines of evidence exist to support essentiality of impaired proteostasis, a single molecular chain of events cannot be established. |
||
|
KE4 Degeneration of DA neurons of nigrostriatal pathway |
MODERATE |
Receptors for advanced glycated end product (AGEs) can activate NF-kB (a transcription factor involved in the inflammatory response) and they are found on microglia cells and astrocytes. Ablation of receptor for advanced glycated end product (RAGE) proved to be protective against MPTP-induced decreases of TH+ neurons and mitigation of microglia and astrocytes reactivity was observed (Teismann et al. 2012). Inhibition of RAGE, which is upregulated in the striatum following rotenone exposure and in response to neuroinflammation, decreases rotenone-induced apoptosis by suppressing NF(Nuclear Factor)-kB activation, as well as the downstream inflammatory markers TNF-alpha, iNOS and myeloperoxidase (Abdelsalam and Safar, 2015). This showed intermingled links between neuronal injury/death and neuroinflammation. Rotenone-induced neurotoxicity was less pronounced in neuron-enriched cultures than in neuron-glia co-cultures (Gao et al., 2002), suggesting that neuron-glia interactions are critical for rotenone-induced neurodegeneration. In addition, in in vitro systems, a decrease in thyroxine hydrosilase (TH) mRNA expression has been observed to be a sufficient signal to trigger microglial reactivity (Sandström et al., 2017). |
||
|
KE5 Neuroinflammation |
MODERATE |
Rationale: Following treatment with Rotenone or MPTP/ MPP+, protection of DA neurons and terminals was observed in vivo and in vitro by inhibiting different feature of neuroinflammation (microglia/astrocyte); however, inhibition was different in different models and considered as an indirect evidence of essentiality (Zhou et al., 2007; Gao et al., 2002 and 2003 and 2015; ; Emmrich et al., 2013; Salama et al., 2012; Chang et al., 2013; Wang et al., 2014; Liu et al., 2012, 2015; Borrajo et al., 2013; Brzozowski et al., 2015; Wang et al., 2006; Chung et al., 2011; Sriram et al., 2014; Feng et al., 2002; Sathe et al., 2012; Khan et al., 2014; Ros-Bernal et al., 2011; Ferger et al., 2004; Chao et al., 2009; Rojo et al., 2010; Qian et al., 2011; Dehmer et al., 2000; Bodea et al., 2014). Mice lacking the type-1 Interferons receptor showed an attenuated pro-inflammatory response and reduced loss of dopaminergic neurons induced by MPTP/MPP+. The neuro-protective potential was also confirmed by treatment with a blocking monoclonal antibody against type-1A IFN receptor (interferon receptor) that increased survival of dopaminergic neurons of TH+ (Main et al., 2016). |
||
|
KE4 Degeneration of DA neurons of nigrostriatal pathway |
STRONG |
Rationale: Clinical and experimental evidences show that the pharmacological replacement of the dopamine (DA) neurofunction by allografting fetal ventral mesencephalic tissues is successfully replacing degenerated DA neurons resulting in the total reversibility of motor deficit in animal model and partial effect is observed in human patient for PD (Widner et al., 1992; Henderson et al., 1991; Lopez-Lozano et al., 1991; Freed et al., 1990; Peschanski et al., 1994; Spencer et al., 1992). Also, administration of L-DOPA or DA agonists results in an improvement of motor deficits (Calne et al 1970; Fornai et al. 2005). The success of these therapies in man as well as in experimental animal models clearly confirms the causal role of dopamine depletion for PD motor symptoms ( Connolly et al., 2014; Lang et al., 1998; Silva et al., 1997; Cotzias et al., 1969; Uitti et al., 1996; Ferrari-Tonielli et al., 2008; Kelly et al., 1987; Walter et al., 2004; Narabayashi et al., 1984; Matsumoto et al., 1976; De Bie et al., 1999; Uitti et al., 1997; Scott et al., 1998; Moldovan et al., 2015; Deuschl et al., 2006; Fasano et al., 2010; Castrito et al., 2011; Liu et al., 2014; Widner et al., 1992; Henderson et al., 1991; Lopez-Lozano et al., 1991; Freed et al., 1990; Peschanski et al., 1994; Spencer et al., 1992). Furthermore, experimental evidence from animal models of PD and from in-vitro systems indicate that prevention of apoptosis through ablation of BCL-2 family genes prevents or attenuates neurodegeneration of DA neurons (Offen D et al., 1998; Dietz GPH et al. 2002). |
||
Evidence Assessment
Concordance of dose-response relationship.
Data from experiments with the stressor compounds rotenone and MPTP (known inhibitors of the mitochondrial Complex I (CI)) reveal a good concordance of the dose-response relationships between the MIE and AO and within KEs. Although the different KEs have been measured using different methodologies, comparison of data from multiple in-vitro/in-vivo studies shows a general agreement in dose-relationship (see table 1 and 2). There is a good consistency when comparing data on KE4 and the AO after exposure to rotenone and MPTP. However, in vivo rodent studies proved that only exposure to low concentrations of rotenone (rat brain concentration between 20-30 nM of rotenone; Betrabet et al., 2000) or MPTP (mice striatum concentration of approximately 12-47 µM MPP+; Fornai et al., 2005; Thomas et al. 2012) after chronic exposure (approximately 5 weeks) reproduced the anatomical, neurochemical behavioural and neuropathological features similar to the ones observed in Parkinson’s disease (PD). Because of the variability of experimental protocols used, a clear no-effect threshold could not be established; nevertheless, these brain concentrations of rotenone (20-30 nM) and MPP+ (approximately 12-47µM) could serve as probabilistic thresholds for chronic exposure that could reproduce features of PD as both concentrations trigger approximately a 50% inhibition of Complex I (see table 3). Generally, a strong response-response relationship is observed within studies. Some exceptions for this rule are observed between KE3/KE5 and KE4, likely because of the all biological complexity associated with these KEs. In this AOP, neuroinflammation was considered to have a direct effect on degeneration of DA neurons. However, it was not clear at which conditions it would become a modulatory factor and for practical reasons was not included in table 1, 2 and 3 but considered in the weight of evidence analysis.
Table1 Dose-response and temporality table for rotenone.
|
Rotenone Concentration |
KE1aaa Inhibition of C I |
KE2aaa Mitochondrial dysfunction |
KE3aaa Impaired proteostasis |
KE4 Degeneration of DA neurons of nigrostriatal pathway |
AO Parkinsonian motor symptoms |
|
5-10 nM in-vitro [1] |
+ 4-72 hours [1] |
+ 4-72 hours [4] |
+ 24 hours [3] |
- |
- |
|
20-30 nM ex-vivo, rat brain concentration [4-5-2-6] |
++ 4-72 hours (4-5) |
++ 4-72 hours [4-5] |
++ 24 hours [3-2-6] |
++a 5 weeks [2-6] |
+++aa 5 weeks [2-6] |
|
100 nM in-vitro [4] |
+++ 4-72 hours [4] |
+++ 4-72 hours [4] |
+++ 24 hours [3] |
Corresponding to a concentration above the maximum tolerated dose in-vivo [2-6] |
Corresponding to a concentration above the maximum tolerated dose in vivo [2-6] |
References: Choi et al. 2008 [1]; Betarbet et al. 2006 [2]; Chou et al. 2010 [3]; Barrientos and Moraes 1999 [4]; Okun et al. 1999 [5]; Betarbet et al. 2000 [6]
-no data available
+: low severity score, ++ intermediate severity score, +++ high severity score
a: 50% of treated animals showed loss of DA neurons in SNpc
aa: All animals affected in KE4 showed impaired motor symptoms
aaa: KE 1, 2 and 3 showed a dose-related severity in the effect and the score ++ was normalized vs. the KE4
Table 2. Dose-Response and Temporality table for MPTP/MPP+
|
MPTP Administered Dose |
MPP+ Brain Concentration |
KE1bb Inhibition of C I |
KE2bb Mitochondrial dysfunction |
KE3b Impaired proteostasis |
KE4 Degeneration of DA neurons of nigrostriatal pathway |
AO Parkinsonian motor symptoms |
|
1 mg/kg sc infusion [1] |
- |
- |
- |
+ 4 weeks[1] |
+aaa 4 weeks [1] |
No effect |
|
5 mg/kg sc infusion [1] |
- |
- |
- |
++ 4 weeks[1] |
++aa 4 weeks [1] |
+++ 4 weeks [1] |
|
20-30 mg/kg sporadic ip. injections (4 times every 2 hours) [2, 1]
|
47µM [2]^ 12µM [1] |
+++ 4 hrs [2] |
+++ 4hrs [2] |
+++ 4 weeks [1] |
+++a 1-4 weeks[2,1]
|
+++ 4 weeks [1] |
References. Fornai et al. 2005 [1]; Thomas et al. 2012 [2]
-no data available
a: approx 50% loss of DA neurons in SNpc
aa: approx 30% loss of DA neurons SN pc
aaa: no loss of DA neurons in SN pc. Reduced level of striata DA
b: for KE3, a dose response effect was observed.
bb: for KE 1 and 2 the severity of the effect was normalized vs. the KE4
^ After single dose MPTP administration, brain concentration was approx. 5.15 µM
Temporal concordance among the MIE, KEs and AO.
There is a strong agreement that loss of DA neurons of the SNpc that project into the putamen is preceded by reduction in DA and degeneration of DA neuronal terminals in the striatum (Bernheimer et al. 1973). The clinical symptoms of a motor deficit are observed when 80% of striatal DA is depleted (Koller et al. 1992) and the sequence of pathological events leading to the adverse outcome has been well-documented (Fujita, et al.2014; O’Malley 2010, Dexter et al. 2013). Temporal concordance (see table 1 and 2) among the KEs can be observed in the experimental models of PD using the chemical stressors rotenone and MPTP (Betarbet 2000 and 2006; Sherer et al. 2003, Fornai et al. 2005). The acute administration of the chemical stressors can trigger a dose-related change from the MIE to impaired proteostasis; however, to trigger KE4 (i.e. degeneration of DA neurons in SNpc with presence of intracytoplasmatic Lewy-like bodies) and motor deficits (AO), proteostasis needs to be disturbed for a minimum period of time (Fornai et al. 2005).
Strength, consistency, and specificity of association of AO and MIE.
Strength and consistency of the association of the AO with the MIE is strong. There is a large body of evidence from in-vitro and in-vivo studies with chemical stressors, showing association between the MIE that triggers an inhibition of CI and the AO (Sherer et al. 2003; Betarbet et al. 2000 and 2006, Fornai et al. 2005). Human data also suggest a link between inhibition of CI and AO (Greenamyre et al. 2001; Schapira et al. 1989; Shults, 2004). Using the two different chemical stressors, rotenone and MPTP, data are consistent and the pattern of activation of the MIE leading of the AO is similar. For rotenone and MPTP, specificity is high; however, there are many inhibitors of the mitochondrial CI without evidence of triggering the AO. When considering the limited amount of chemical stressors for which empirical data are available for supporting the full sequence of KEs, kinetic and metabolic considerations should be taken into account to demonstrate specificity for these compounds. The issue of specificity was also debated during the external review of this AOP and the following information was added:
The vast majority of empirical support available in the literature is based on complex I inhibitors, such as rotenone and MPTP/MPP+, as well as on studies involving genetic impairment of complex I activity. A relatively wide spectrum of structurally different complex I inhibitors have been described over the course of recent decades. Prominent examples are acetogenins (Nat Prod Rep 2005, 22, 269-303); alkaloids (J Neurochem 1996, 66, 1174-1181); antibiotics (BBA 1998, 1364, 222-235; Eur J Biochem 1994, 219, 691-698; JBC 1970, 245, 1992-1997; Bioorg Med Chem 2003, 11, 4569-4575); pesticides (Biochem Soc Trans 1994, 22, 230-233); quinones (JBC 1971, 246, 2346-2353); or vanilloids (ABB 1989, 270, 573-577). Additional information can be also retreived from Fato et al 2009, Espositi et al. 1993, Lagoa et al. 2011 and Park et al. 2003.
All of these structurally different complex I inhibitors were characterized with isolated mitochondria or with submitochondrial particles. Application of bovine heart mitochondria revealed IC50 values in the range of 20-70 nM for piericidin A, fenpyroximate, rotenone, and phenoxan (Eur. J. Biochem 1994, 219, 691-698). IC50 values in the range of 1-10 nM were detected by application of submitochondrial particles with rotenone, molvizarin, rollinstatin-1 and -2, and piericidin A (Biochem J. 1994, 301, 161-167).
Studies involving neuronal cell cultures or in vivo models are in fact rather rare. A systematic comparison of the IC50 values for complex I inhibition and EC50 values for the reduction of ATP levels; cell death was performed with rat fetal striatal neurons (Exp Neurol 2009, 220, 133-142). Due to the lipophilicity of most of the complex I inhibitors tested, the detected EC50 values were in most cases lower than the IC50 values detected for complex I inhibition. EC50 values detected were: annonacin (60 nM), fenazaquin (45 nM), piericidin A (1.6 nM), rollinstatin- 2 (1 nM), rotenone (8 nM), and squamocin (1 nM).
A systematic investigation involving mesencephalic cultures as well as rats was performed for the complex I inhibitor annonacin, a major acetogenin of soursop, a plant suspected to cause an atypical form of PD in Guadeloupe. Mesencephalic cultures treated for 24 h with annonacin revealed EC50 values of 20 nM (annonacin), 34 nM (rotenone), and 1900 nM (MPP+) (Neurosci 2003, 121(2), 287-296). Intravenous application by minipumps over the course of 28 days indicated a passage of annonacin across the blood-brain barrier, and an energy-dependent loss of ca. 30 % of DA neurons in the substantia nigra (Champi et al.2004)).
Weight of Evidence (WoE).
Biological plausibility, coherence, and consistency of the experimental evidence.
The biological plausibility of this AOP is overall considered strong. When using multiple stressors in different studies and assays, the coherence and consistency of the experimental data is well established. Furthermore, in-vivo and in-vitro studies are also in line with the human evidence from PD patients. In addition, although the mechanistic understanding of parkinsonian disorders (and PD in particular) are not fully clear, the KEs and KERs described in this AOP are considered critical for the development of the disease (Fujita et al. 2015, Shulman et al. 2011, Dexter et al. 2013, Dauer et al. 2003).
|
1 Support for Biological Plausibility of KERs |
Defining Question |
High (Strong) |
Moderate |
Low(Weak) |
|
Is there a mechanistic (i.e. structural or functional) relationship between KEup and KE down consistent with established biological knowledge? |
Extensive understanding of the KER based on extensive previous documentation and broad acceptance |
The KER is plausible based on analogy to accepted biological relationships, but scientific understanding is not completely established |
There is empirical support for a statistical association between KEs but the structural or functional relationship between them is not understood |
|
|
MIE => KE1 Binding of inhibitor to NADH-ubiquinone oxidoreductase leads of complex I |
STRONG |
Rationale: As describe in this KER there is an extensive understanding of the functional relationship between binding of an inhibitor to NADH-ubiquinone oxidoreductase (CI) and its inhibition. Different complex I ligands, both naturally occurring, like rotenone (from Derris scandens), piericidin A (from Streptomyces mobaraensis), acetogenins (from various Annonaceae species) and their derivatives, and synthetically manufactured like pyridaben and various piperazin derivatives inhibit the catalytic activity of complex I (Degli Esposti, 1994: Ichimaru et al. 2008; Barrientos and Moraes, 1999; Betarbet et al., 2000). |
||
|
KE1 => KE2 Inhibition of complex I leads to mitochondrial dysfunction |
STRONG |
Rationale: There is extensive understanding of the mechanisms explaining how the inhibition of complex I lead to mitochondrial dysfunction (i.e. failure to produce ATP, increase in production of ROS etc). It is well documented that CI inhibition is one of the main sites at which electron leakage to oxygen occurs resulting in oxidative stress (Efremov and Sazanow, 2011; lauren et al. 2010; Greenamyre et al. 2001). These pathological mechanisms are well studied as they are used as readouts for evaluation of mitochondrial dysfunction (Graier et al., 2007; Braun, 2012; Martin, 2011; Correia et al., 2012; Cozzolino et al., 2013 |
||
|
KE2 => KE3 Mitochondrial dysfunction results in impaired proteostasis |
MODERATE |
Rationale: The weight of evidence supporting the biological plausibility behind the relationship between mitochondrial dysfunction and impaired proteostasis, including the impaired function of UPS and ALP that results in decreased protein degradation and increase protein aggregation is well documented but not fully understood. It is well established that the two main mechanisms that normally remove abnormal proteins (UPS and ALP) rely on physiological mitochondrial function. The role of oxidative stress, due to mitochondrial dysfunction, burdens the proteostasis with oxidized proteins and impairs the chaperone and the degradation systems. This leads to a vicious circle of oxidative stress inducing further mitochondrial impairment (Powers et al., 2009; Zaltieri et al., 2015; McNaught and Jenner, 2001). Therefore, the interaction of mitochondrial dysfunction and UPS /ALP deregulation plays a pivotal role in the pathogenesis of PD (Dagda et al., 2013; Pan et al., 2008; Fornai et al., 2005; Sherer et al., 2002). |
||
|
KE2 => KE4 Mitochondrial dysfunction leads to the degeneration of dopaminergic neurons of the nigrostriatal pathway |
STRONG |
Rationale: Mitochondrial are essential for ATP production, ROS management, calcium homeostasis and control of apoptosis. Mitochondrial homeostasis by mitophagy is also an essential process for cellular maintenance (Fujita et al. 2014). Because of their anatomical and physiological characteristics, SNpc DA neurons are considered more vulnerable than other neuronal populations (Sulzer et al. 2013; Surmeier et al.2010). Mechanistic evidence of mutated proteins relate the mitochondrial dysfunction in familial PD with reduced calcium capacity, increased ROS production, increase in mitochondrial membrane permeabilization and increase in cell vulnerability (Koopman et al. 2012; Gandhi et al. 2009). Human studies indicate mitochondrial dysfunction in human idiopathic PD cases in the substantia nigra (Keeney et al., 2006; Parker et al., 1989, 2008; Swerdlow et al., 1996). In addition, systemic application of mitochondrial neurotoxicants such as rotenone or MPTP leads to a preferential loss of nigrostriatal DA neurons (Langston et al., 1983). |
||
|
KE3 => KE4 Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway |
MODERATE |
Rationale: It is well known that impaired proteostasis refers to misfolded and aggregated proteins including alfa-synuclein, deregulated axonal transport of mitochondria and impaired trafficking of cellular organelles. Evidences are linked to PD and experimental PD models as well as from genetic studies (McNaught et al. 2001, 2003; Tieu et al. 2014; Arnold 2011; Rappold et al. 2014). Strong evidence for degeneration of the nigrostriatal pathway comes from the experimental manipulations that directly induce the same disturbances of proteostasis as observed in PD patients (e.g. viral mutated alpha-synuclein expression) or in chronic rotenone/MPTP models trigger degeneration of the nigrostriatal pathway (Kirk et al. 2003; Betarbet et al. 2000 and 2006; Fornai et al. 2005). However, a clear mechanistic proof for the understanding of the exact event triggering cell death is lacking. There is only moderate evidence showing that interventions that correct disturbances of proteostasis after exposure to rotenone would prevent neuronal degeneration and that the disturbances of proteostasis correlate quantitatively under many conditions with the extent of nigrostriatal neuronal degeneration. |
||
|
KE4 => KE5 Degeneration of DA neurons of the nigrostriatal pathway leads to neuroinflammation |
MODERATE |
Rationale: The fact that neuronal injury/death can trigger neuroinflammation is supported by evidence in human and experimental models. The evidence that neuroinflammation triggered by neuronal damage can cause neuronal death (vicious circle), is mostly indirect (blockade of any feature of neuroinflammation) or by analogy (Hirsch and Hunot, 2009; Tansey and Goldberg, 2009; Griffin et al., 1998; McGeer and Mc Geer, 1998; Blasko et al., 2004; Cacquevel et al., 2004; Rubio-Perez and Morillas-Ruiz, 2012; Thundyil and Lim, 2014; Barbeito et al., 2010). Neuroinflammation is observed in idiopathic and in genetic human PD as well as in complex I inhibitor exposed humans, non-human primates, and rodent. Components of damaged neurons lead to glial cells activation via Toll-like receptors. Several chemokines and chemokine receptors (fraktalkine, CD200) control the neuron-microglia interactions. Neuroinflammation in response to damaged neurons is not confined to PD, but is common to several neurodegenerative diseases |
||
|
KE5 => KE4 Neuroinflammation leads to degeneration of DA neurons of the nigrostriatal pathway |
MODERATE |
Rationale: The fact that reactive glial cells (microglia and astrocytes) may kill neurons is well accepted. The mechanisms underlying this effect may include the release of cytotoxic signals (e.g. cytokines) or production of ROS and RNS (Chao et al., 1995 ; Brown and Bal-Price, 2003 ; Kraft and Harry, 2011 ; Taetzsch and Block, 2013). However, the responsible mediators differ from model to model. In humans or non-human primates, an inflammatory activation of glial cells is observed years after exposure to complex I inhibitors. Activated microglia and astrocytes form pro-inflammatory cytokines and free radical species, mostly responsible for neuronal damage. Glial reactivity promotes an impairment of blood brain barrier integrity, allowing an infiltration of peripheral leukocytes that exacerbate the neuroinflammatory process and contribute to neurodegeneration.The debris of degenerating neurons causes neuroinflammation, which in turn can trigger neurodegeneration, thus leading to a self-perpetuating vicious cycle. |
||
|
KE4 => AO Degeneration of DA neurons of the nigrostriatal pathway leads to parkinsonian motor symptoms |
STRONG |
Rationale: The mechanistic understanding of the regulatory role of striatal DA in the extrapyramidal motor control system is well established. The loss of DA in the striatum is characteristic of all aetiologies of PD and is not observed in other neurodegenerative diseases (Bernheimer et al. 1973; Reynolds et al. 1986). Characteristic motor symptoms such as bradykinesia, tremor, or rigidity are manifested when more than 80 % of striatal DA is depleted as a consequence of SNpc DA neuronal degeneration (Koller et al. 1992). |
||
Empirical support.
Empirical support is strong. Many studies show evidence for the KERs by showing temporal concordance and dose concordance when using different stressors.
|
3 Empirical support for KERs |
Defining Question Does the empirical evidence support that a change in the KEup leads to an appropriate change in the KE down? Does KEup occur at lower doses and earlier time points than KE down and is the incidence of KEup higher than that for KE down? Are inconsistencies in empirical support cross taxa, species and stressors that don’t align with expected pattern of hypothesized AOP? |
High (Strong) |
Moderate |
Low(Weak) |
|
Multiple studies showing dependent change in both exposure to a wide range of specific stressors (extensive evidence for temporal, dose-response and incidence concordance) and no or few critical data gaps or conflicting data. |
Demonstrated dependent change in both events following exposure to a small number of specific stressors and some evidence inconsistent with expected pattern that can be explained by factors such as experimental design, technical considerations, differences among laboratories, etc. |
Limited or no studies reporting dependent change in both events following exposure to a specific stressor (ie endpoints never measured in the same study or not at all); and/or significant inconsistencies in empirical support across taxa and species that don’t align with expected pattern for hypothesized AOP |
||
|
MIE => KE1 Binding of inhibitor to NADH-ubiquinone oxidoreductase leads to partial or total inhibition of complex I |
STRONG |
Rationale: The inhibition of complex I is well documented in a variety of studies using isolated mitochondria or cells as well as in in vivo experiments and in human post mortem PD brains. In many experiments using different inhibitors ie rotenone and MPTP, the observed relationship between the two events was temporal, response and dose concordant (Betarbet et al., 2000 and 2006, Okun et al., 1999, Koopman et al., 2007, Choi et al., 2008, Grivennikova et al., 1997, Barrientos and Moraes 1999). |
||
|
KE1 => KE2 Inhibition of complex I leads to mitochondrial dysfunction |
STRONG |
Rationale: There is a large amount of studies showing that the inhibition of CI inhibition results in mitochondrial dysfunctions in a response and dose dependent manner (Barriento and Moraes, 1999). |
||
|
KE2 => KE3 Mitochondrial dysfunction results in impaired proteostasis |
STRONG |
Rationale: Based on the existing in vitro and in vivo data it is suggested that mitochondrial dysfunction impairs protein homeostasis (impairment of the UPS and ALP system) through oxidative and nitrosative stress resulting in accumulation of misfolded proteins (including α-synuclein), disruption of microtubule assembly and damaged intracellular transport of proteins and cell organelles. A number of studies performed with chemical stressors showed evidence of temporal, response and dose concordance (Chou et al. 2010; Betarbet et al. 2000 and 2006; Fornai et al. 2005). |
||
|
KE2 => KE4 Mitochondrial dysfunction directly leads to degeneration of DA neurons of nigrostriatal pathway |
STRONG |
Rationale: Multiple in vitro studies indicate dose and response-response concordance. As most of the studies were conducted in vitro, the temporal concordance is difficult to establish; however, can be expected based on the well know temporal sequence of the two KEs. (Park et al., 2014; Choi et al., 2014; Marella et al., 2008; Du et al. 2010; Hajieva et al., 2009; Sherer et al., 2003; Sherer et al., 2007; Wen et al. 2011; Swedlow et al., 1996; Jana et al., 2011; Jha et al., 2000; Chinta et al., 2006) |
||
|
KE3 => KE4a Impaired proteostasis leads to degeneration of DA neurons of the nigrostriatal pathway |
STRONG |
Rationale: The empirical support linking impaired proteostasis with degeneration of DA neurons of the nigrostriatal pathway is strong and comes from in-vivo and in-vitro studies performed with different stressor (i.e. Rotenone, MPTP or proteasome inhibitors) and post-mortem human evidences in PD patients supporting a causative link between the two key events. Temporal, effect and dose concordance was established in a number of experiments (Fornai et al. 2005; Fornai et al. 2003; Betabret et al. 2000 and 2006). |
||
|
KE4a => KE5 Degeneration of DA neurons of nigrostriatal pathway leads to neuroinflammation |
MODERATE |
Rationale: multiple in vivo and in vitro experiments support the link between degeneration of DA neurons in the nigrostriatal pathway and neuroinflammation. The observation of concomitant presence of reactive microglial and astrocytic cells and degenerated/degenerating DA neurons is also reported in many studies with a good temporal and response concordance. ATP and other damage associated molecular patterns (DAMPs), released from degenerating cells, stimulate P2Y receptors on microglia, leading to their activation. Experimental injection of DAMPs, fraktalkine, or neuromelanin, released by degenerating DA neurons evokes neuroinflammation. Neutralization of DAMPs (e.g. antibodies against HMGB1 or CX3CR1) decreases MPTP-induced neuroinflammation. Toll-like receptor 4 deficient mice display a reduced neuroinflammatory response upon MPTP treatment. Inhibition of RAGE, which is upregulated in striatum upon rotenone exposure, suppresses NF-kB activation and downstream inflammatory markers. |
||
|
KE5 => KE4b Neuroinflammation leads to degeneration of DA neurons of nigrostriatal pathway. |
MODERATE |
Rationale: multiple in vivo and in vitro experiments support the link between neuroinflammation and degeneration of DA neurons in the nigrostriatal pathway. The observation of concomitant presence of reactive microglial and astrocytic cells and degenerated/degenerating DA neurons is also reported in many studies with a good temporal and response concordance. Neuroinflammation has been implicated in dopaminergic neuronal cell death in PD patients (Vivekanantham et al., 2014). LPS injection into the CNS, or applied systemically, evokes glial inflammation and a preferential degeneration of DA neurons. In mouse models with a knockout of either IL-1b, IFN-g, or TNF-a receptors 1 and 2, LPS no longer evokes neuroinflammation and DA neurodegeneration. Experimental interference with CD4+ T cell activation protects from DA neurodegeneration. Transfer of immunosuppressive regulatory T cells protect from DA neurodegeneration. Anti-inflammatory TGF-b1 signaling protects from DA neurodegeneration. Clinical trials indicate a protective influence on DA neuron survival by the antibiotic minocycline blocking microglial reactivity, in association with rasagiline (prevents DA degeneration), and coenzyme Q10/creatine (restoration of cellular ATP). |
||
|
KE4b => AO Degeneration of DA neurons of nigrostriatal pathway leads to parkinsonian motor symptoms |
STRONG |
Rationale: The experimental support linking the degeneration of DA neurons of nigrostriatal pathways with the manifestation of motor symptoms of PD comes from human in vivo observations as well as from monkey, mice and rat in vivo models exposed to an experimental toxin i.e. rotenone and MPTP. Observations in human allow defining correlation between the levels of striatal DA with the onset of motor dysfunction (Lloyd et al. 1975; Hornykiewicz et al. 1986; Bernheimer et al. 1973). Temporal, effect and dose concordance comes from studies performed in multiple animal species following administration of rotenone and MPTP (Bezard et al. 2001; Cannon et al. 2009; Petroske et al. 2001; Alvarez-Fischer et al. 2008; Blesa et al. 2012; Lloyd et a. 1975). |
||
Uncertainties and Inconsistencies.
- The strength of this AOP is mainly based on MPP+ and rotenone and only limited information on whether other mitochondrial complex I inhibitors also perturb the KEs (specifically degeneration of DA neurons in the SNpc) or produce a similar AO.
- Conflicting data exists (Choi et al. 2008) showing that mitochondrial complex I inhibition is not required for DA neuron death induced by rotenone, MPTP/MPP+, or paraquat, challenging the current AOP. The cited research article shows that abolishment of complex I’s activity by inactivation of a gene that codes for a subunit of complex I does not impact the survival of DA neurons in culture. The actions of rotenone, MPTP/MMP+ are independent of complex I. Since some complex I inhibitors also target other complexes, it is possible that impairment of other respiratory complexes may be involved. It was noted that this paper used the approach of genetically deleting an essential chaperone in complex I assembly, and the authors found that absence of complex I activity in this model did not affect the toxicity of rotenone and MPP+. However, the findings have never been confirmed/ continued, neither in the originating laboratory, nor by others. Second, the work did not consider the possibility that some functions of complex I were not affected by the absence of the chaperone (e.g. reverse electron transfer from complex II and III), and that rotenone and MPTP/MPP+ may well cause toxicity by interfering with such residual function (e.g. by favoring channeling of electrons to molecular oxygen). In light of this situation, the publication of Choi et al (2008) should be considered weak in the overall weight of evidence and therefore considered a minor inconsistency.
- There is no strict linear relationship between inhibitor binding and reduced mitochondrial function. Low doses of rotenone that inhibit CI activity partially do not alter mitochondrial oxygen consumption. Therefore, bioenergetics defect cannot account alone for rotenone-induced neurodegeneration. Instead, under such conditions, rotenone neurotoxicity may result from oxidative stress (Betarbet et al., 2000). Few studies used human brain cells/human brain mitochondria. Therefore, full quantitative data for humans are not available.
- It is molecularly unclear how rotenone binding alter CI function, switching it to ROS production. It is still unclear whether the site of superoxide production in CI inhibited mitochondria is complex I itself or not (Singer and Ramsay, 1994).
- Some studies suggest that rotenone and MPTP/MPP+ may have effects other than CI inhibition, e.g. MPTP and rotenone can induce microtubule disruption (Feng, 2006; Ren et al., 2005; Cappelletti et al., 1999; Cappelletti et al., 2001, Brinkley et al., 1974; Aguilar et al., 2015).
- There are additional feedback possible between KEs, e.g. ROS production from KE2 may damage CI, this leads to enhancement of KE1.
- Some KEs e.g. KE 2, 3, 5 pool molecular processes that may need to be evaluated individually at a later stage.
- The exact molecular link from mitochondrial dysfunction to disturbed proteostasis is still unclear (Malkus et al., 2009; Zaltieri et al., 2015).
- The role of ATP depletion vs. other features of mitochondrial dysfunction is not clear.
- The role of a α-synuclein in neuronal degeneration is still unclear as well as the mechanisms leading to its aggregation.
- It is not clear under which conditions KE3 and KE5 become modulatory factors, and when they are essential. MPTP/MPP+ can induce damage to nigrostriatal neurons without formation of Lewy bodies (Dauer 2003; Forno 1986, 1993). Similarly, discontinuous administration of rotenone, even at high doses, damages the basal ganglia but produce no inclusions (Heikkila et al., 1985; Ferrante et al., 1997, Lapontine 2004). To reproduce the formation of neuronal inclusions, continuous sc infusion of MPTP/MPP+ or rotenone is necessary. Acute intoxication with rotenone seems to spare dopaminergic neurons (Dauer et al., 2003, Ferrante 1997). In addition, in rats chronically infused with rotenone showed a reduction in striatal DARPP-32-positive (dopamine- and cyclic-AMP-regulated phosphoprotein of molecular weight 32,000), cholinergic and NADPH diaphorase-positive neurons (Hoglinger 2003) or in other brain regions. These results would suggest that Rotenone can induce a more widespread neurotoxicity (Aguilar 2015) or the model is not reproducible in all laboratories.
- Inconsistent effects of MPTP/MPP+ on autophagy (up or down regulation) are reported (Drolet et al., 2004: Dauer et al., 2002). There is conflicting literature on whether increased autophagy would be protective or enhances damage. Similarly, a conflicting literature exists on extent of inhibition or activation of different protein degradation system in PD and a clear threshold of onset is unknown (Malkus et al., 2009; Fornai et al., 2005).
- The selective vulnerability of the SNpc dopaminergic pathway does not have a molecular explanation.
-
In some instances, the differential vulnerability of various brain regions towards a generalized complex I inhibition found non-dopaminergic lesions, particularly in the striatum, in all animals with nigral lesion, as seen in atypical parkinsonism but not in idiopathic Parkinson's disease (Hoglinger et al., 2003)
- Priority of the pattern leading to cell death could depend on concentration, time of exposure and species sensitivity; these factors have to be taken into consideration for the interpretation of the study’s result and extrapolation of potential low-dose chronic effect as this AOP refers to long-time exposure.
- The model of striatal DA loss and its influence on motor output ganglia does not allow to explain specific motor abnormalities observed in PD (e.g. resting tremor vs bradykinesia) (Obeso et al., 2000). Other neurotransmitters (Ach) may play additional roles. Transfer to animal models of PD symptoms is also representing an uncertainties.
- There are some reports indicating that in subacute rotenone or MPTP models (non-human primates), a significant, sometimes complete, recovery of motor deficits can be observed after termination of toxicant treatment. The role of neuronal plasticity in intoxication recovery and resilience is unclear.
- This AOP is a linear sequence of KEs. However, mitochondrial dysfunction (and oxidative stress) and impaired proteostasis are influencing each other and this is considered an uncertainties (Malkus et al., 2009).
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
Concentration-response data for the prototypical stressors deguelin, fenpyroximate, pyrimidifen, rotenone and tebufenpyrad has been derived from Alimohammadi et al. 2023 Delp et al. 2019, 2021, van der Stel-OECD 2020, van der Stel et al. 2020 (Fig. 1). Data points were extracted manually from graphs using PlotDigitizer (v3.0.0). The curve fits were generated using the L.4 function from the drc package in R and organised across KEs to facilitate the analysis of the concordance of the concentration-response relationship. The different KEs have been measured in vitro.
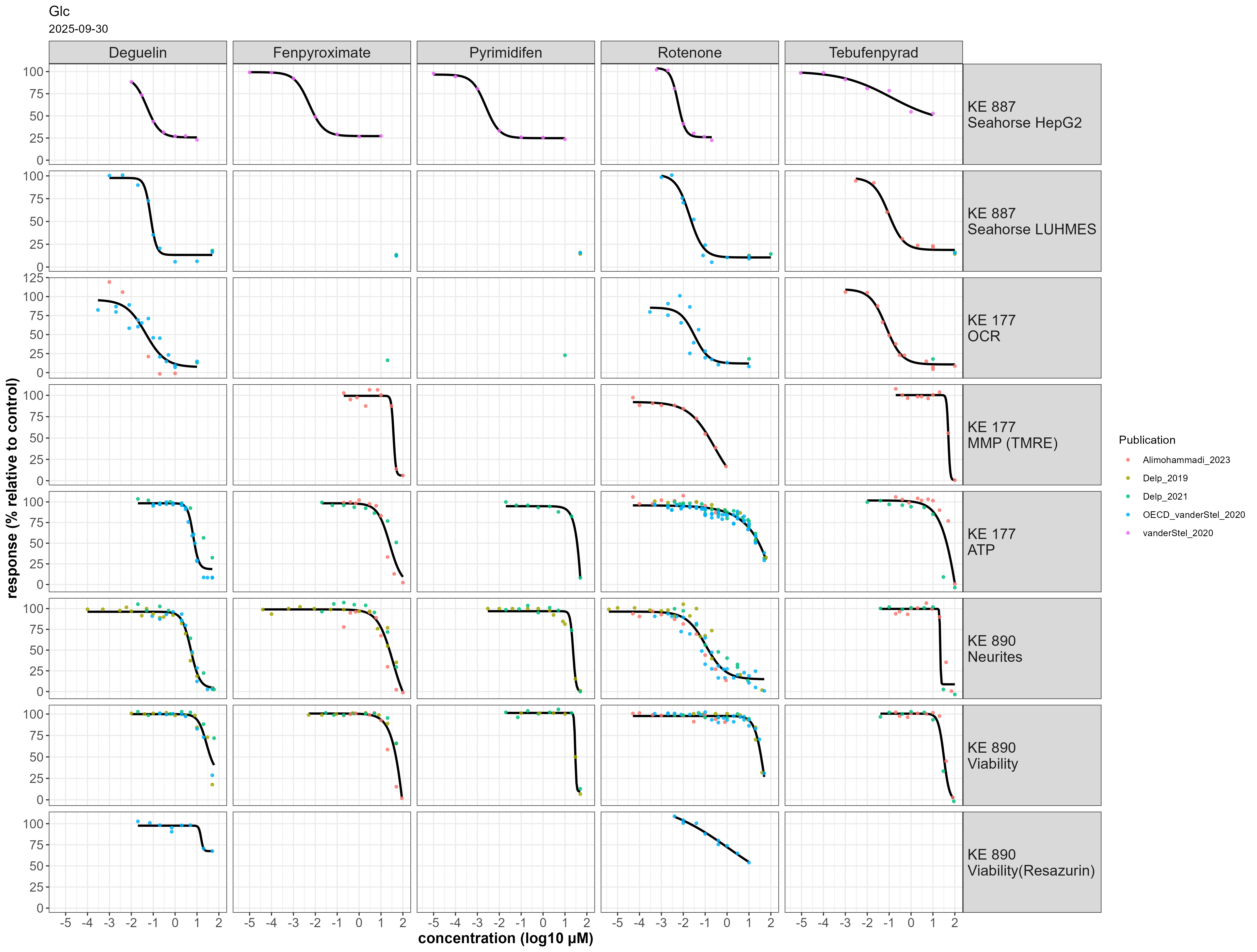 Figure 1: Concentration-response data for the prototypical stressors deguelin, fenpyroximate, pyrimidifen, rotenone and tebufenpyrad across eight endpoints. All experiments were performed using cells cultured in glucose-containing medium.
Figure 1: Concentration-response data for the prototypical stressors deguelin, fenpyroximate, pyrimidifen, rotenone and tebufenpyrad across eight endpoints. All experiments were performed using cells cultured in glucose-containing medium.
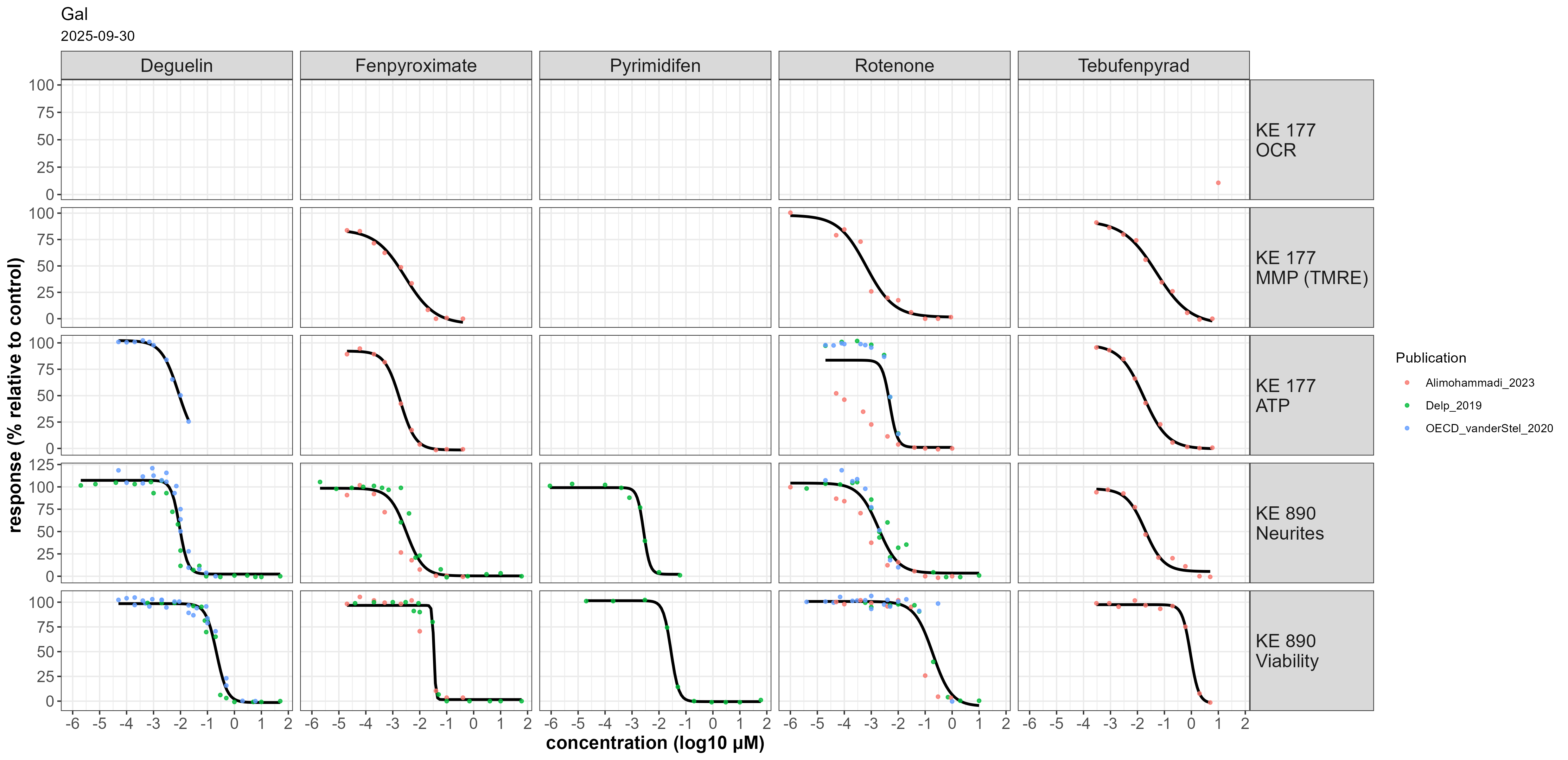 Figure 2: Concentration-response data for the prototypical stressors deguelin, fenpyroximate, pyrimidifen, rotenone and tebufenpyrad across five endpoints with cells cultured in galactose-containng medium.
Figure 2: Concentration-response data for the prototypical stressors deguelin, fenpyroximate, pyrimidifen, rotenone and tebufenpyrad across five endpoints with cells cultured in galactose-containng medium.
Deguelin affected KE 887 in both LUHMES and HepG2 cells at 10-30 nM in permeabilized cells. Oxygen consumption rate was affected at a similar concentration in intact cells (KE 177). ATP content, neurite integrity and viability were affected at ~ 10 µM when cultured in glucose containing medium (Fig 1). When using galactose-containing medium, KE 177 and KE 890 were affected at concentrations similar to KE 887 (Fig2).
Rotenone affected KE 887 in both LUHMES and HepG2 cells at 60-100 nM in permeabilized cells. Oxygen consumption rate and mitochondrial membrane potential was affected at a similar concentration in intact cells (KE 177) (Fig 1). ATP levels and cell viability was inhibited only at higher concentrations (4 – 40 µM) when using glucose-containing medium. Interestingly, neurite integrity was already damaged at 20 – 200 nM, in line with KE 887 and KE 177, suggesting the contribution of additional mechanisms other than cI inhibitors. This is in accordance with studies showing that rotenone inhibits microtubule assembly independently of a specific energy-requiring step (Brinkley et al. 1974; Marshall et al. 1978). This effect is particularly relevant because LUHMES cells are exposed at day 2 of differentiation, an early developmental stage characterised by neurite outgrowth, where microtubules dynamics play a critical role (Rieder et al. 1997, Dehmelt and Shelley)
For cells cultured in galactose-containing medium, nanomolar rotenone concentrations were sufficient to reduce ATP and viability (10-100 nM), with neurite integrity still being more sensitive.
Fenpyroximate inhibited KE 887 in HepG2 at ~ 20 nM; no concentration-response data was available for LUHMES cells. KE 177 and KE 890 were affected at 10 – 50 µM when using glucose-containing medium (Fig 1). When using galactose-containing medium, KE 177 and KE 890 were affected at concentrations similar to KE 888 (Fig 2).
Pyrimidifen inhibited KE 888 in HepG2 at ~7 nM; no concentration-response data was available for LUHMES cells. KE 177 and KE 890 were affected at 16-50 µM when using glucose-containing medium (Fig 1). In galactose-containing medium, only KE 890 was measured, with EC50s for neurite integrity and viability of 3 and 30 nM, respectively (Fig 2).
Tebufenpyrad inhibited KE 888 in LUHMES cells at 40 nM (n = 1 biological replicate) and in HepG2 at 5.5 µM. In intact LUHMES cells cultured in glucose-containing medium, oxygen consumption rate was affected at 45 nM, but mitochondrial membrane potential, ATP content and KE 890 only at 10 – 50 µM (Fig 1). In galactose-containing medium, mitochondrial membrane potential was affected at 6 nM and ATP content between 6-10 nM. EC50s for neurite integrity and viability were 10-40 and 130 – 660 nM, respectively (Fig. 2).
Overall cI inhinibitors show concordance in the concentrations response relationships across KEs
Table 2 Summary of quantitative effects of cI inhibitors
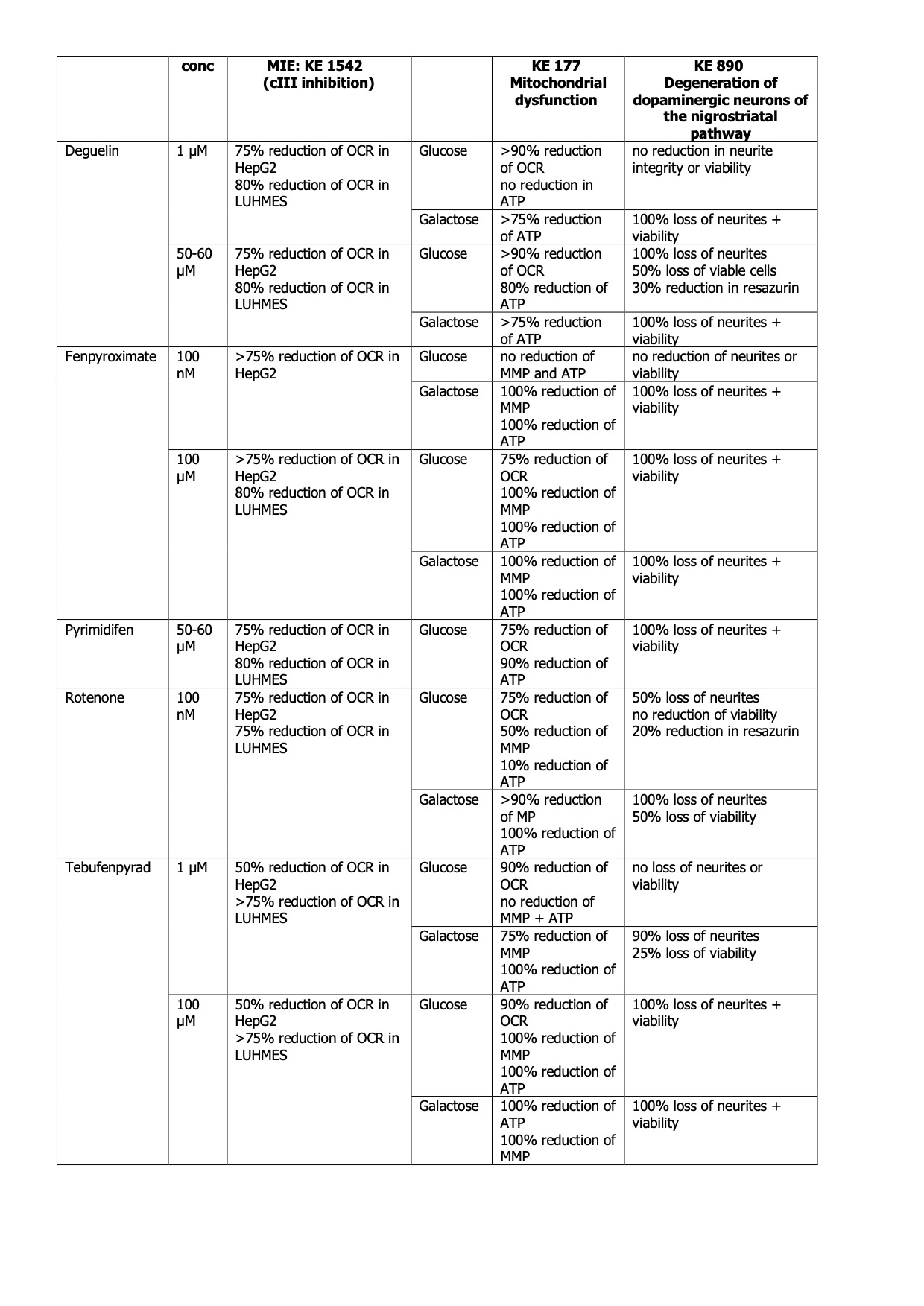
Uncertainties and inconsistencies table
|
Uncertainty |
Impact |
Reason |
|
KE 888 measured in permeabilised cells |
|
Permeabilisation provides direct access for the tested compounds and substrates to the mitochondria and respiratory chain components. The physicochemical properties of the tested compound may reduce its ability to permeate the plasma membrane of intact cells, thus reducing or preventing its uptake which could affect the concentration and the time required to impact the downstream KEs. |
|
Lack of data in galactose condition |
High |
In vitro cell models in general are characterized by an unphysiological reliance on glycolysis. In the presence of glucose any KE is influenced by the contribution of oxidative phosphorylation in addition to glycolisis to meet the cellular need for ATP. Thus, the KEs are influenced by the glycolisis rate. Glucose concentrations in culture medium higher than the physiological level enhances cellular resistance to mitochondrial dysfunction. Application of galactose instead of glucose in the medium allows a shift towards mitochondrial ATP generation. Even under these conditions, glycolysis significantly contributes to ATP production. |
|
Use of HepG2 concentration response curves related to the measurement of oxygen consumption upon inhibition of cIII as a surrogate to represent inhibition of cIII in LUHMES cells, due to the lack of concentration response data for LUHMES cells |
Low |
It is assumed that since the exposure is acute and in permeabilized cells, the test chemical would have immediate access to the mitochondria. Other mechanisms such as transport into the cells or an indirect effect via other signaling pathways were considered negligible under these assay conditions. |
|
Brain vs liver mitochondria |
- |
A study by Balmaceda et al. (2024) in isolated mitochondria provides evidence for intrinsic bioenergetic differences between brain and liver mitochondria obtained from mice, highlighting tissue-specific substrate preferences, redox states, and sensitivity to electron transport system (ETS) deficiencies. Their findings demonstrate that brain mitochondria rely more heavily on Complex I (CI) substrates and exhibit greater vulnerability to CI and Complex III (CIII) dysfunction, whereas liver mitochondria preferentially utilize Complex II (CII) substrates and show metabolic resilience (Balmaceda et al., 2024). These observations are supported by Lesner et al. (2022), who reported that CI is dispensable in liver but essential in brain tissue, and by Szibor et al. (2020), who confirmed higher ROS production in brain mitochondria under reverse electron transport (RET) conditions. Additionally, Rossignol et al. (1999, 2000) emphasized tissue-specific thresholds in oxidative phosphorylation (OXPHOS) control. By contrast, Gusdon et al. (2015) observed similar ETC enzyme activities in mitochondria from different tissues. However, they also confirmed a greater tendency for ROS production in brain mitochondria. This suggests that functional outcomes may be more dependent on systemic or regulatory factors than on the intrinsic properties of mitochondria. The differing results regarding the electron transport system may be due to the higher methodological resolution employed by Balmaceda et al. (2024), which included high-resolution respirometry and real-time coenzyme Q redox monitoring rather than bulk measurements of enzymatic activity and substrate transport. This approach permitted a more detailed and mechanistic understanding of ETS sensitivity and tissue-specific mitochondrial function. Additional factors that can contribute to tissue-specific differences are mtDNA heteroplasmy and lineage-specific transcriptional networks established during development (Burr et al. 2023). |
|
no concentration-response data for OCR in LUHMES |
High |
Increase the uncertainty in the concordance concentration response relationship across the KEs |
|
Exposure is limited in concentrations and to 24 h |
High |
It is possible that the effects on KE 887 and KE177 occur with higher potency or occur more rapidly than those required to observe an effect on KE890. The loss of temporal resolution may determine an excessive steepness of the dose–response curve. |
|
Neurite outgrowth assays (NA)
|
Medium |
Typically conducted from DoD2 to DoD3. NA tested on differentiating neurons is not representative of an adult stage. Active molecular mechanisms that are no longer present in adult or differentiated cells are involved in development. A reduction in NA area may be due to the degeneration of neurites or interference with developing pathways. In this exposure scenario, it is unclear whether the chemical would lead to a loss of neurons, or only a delay in neurite outgrowth. A loss in neurite integrity in the absence of a loss of viability was not considered sufficient to indicate activation of KE 890. |
|
Data reporting |
Medium - Low |
For some assays and chemicals, only two biological replicates were performed (instead of 3), therefore results should be considered with caution (see KERs empyrical evidence). For some assay-chemical combinations it was unclear whether multiple studies repeated the same experiment, or if existing data was reprinted. If experiments were repeated and similar results obtained, this would indicate a higher confidence in the results. If results were simply re-printed, this can lead to an overestimated confidence. As the underlying raw data was not available, it was not possible to investigate further. Different assays have a different effect concentration (i.e. EC25 and EC50). Occasionally, also the same assay can have different effect levels depending on the publication, which reduces overall comparability. However, in most cases the EC25 and EC50 are within a factor of 3 of each other, thus limiting the uncertainty. |
Temporal concordance across the AOP
KE887 is effective withinseconds and changes in MMP (KE177) can be detected within minutes. Time concordance could not be evaluated across KEs 177 and 890 since measurements were available at a single time point (24 h). - Not endorsed
Known Modulating Factors
| Modulating Factor (MF) | Influence or Outcome | KER(s) involved |
|---|---|---|
Quantitative Understanding
The quantitative understanding of this AOP includes a clear response-response relationship and the identification of a threshold effect. The WoE analysis clearly supports the qualitative AOP as a means to identify and characterize the potential of a chemical to induce DA neuronal loss and the AO. Importantly, both the AO and the KE4 are considered relevant regulatory endpoints for this AOP. The empirical evidence supports existence of a response-response relationship. This response-response is likely triggered by a the brain concentrations of approximately 20-30 nM and 17-47 µM of rotenone and MPTP/MPP+ respectively and both concentrations trigger approx. a 50% inhibition of mitochondrial complex I and this could be considered as a “threshold”. However, a more detailed dose-response analysis for each KE is lacking as well as it is not clear which temporal relationship exists for lower CI inhibitory effects. It is clear from the analysis of the AOP that for the identification of these AOs, the design of the in-vivo studies should be tailored as to a MIE which leads to a long-lasting perturbation of the KEs. This provides the most specific and definite context to trigger neuronal death. To observe KEs relevant for this AOP, new endpoints need to be introduced. Although a dose, response and temporal relationship is evident for most KEs, the quantitative relationship between impaired proteostasis and degeneration of DA neurons has yet to be elucidated. Moving from a qualitative AOP to quantitative AOP would need a clear understanding of effect thresholds and this is still representing a major hurdle for several KEs of this AOP.
Table 3 Summary of quantitative effects at the concentration of rotenone and MPTP triggering the AO.
|
Concentration |
KE1 Inhibition of C I |
KE2 Mitochondrial dysfunction |
KE3 Impaired proteostasis |
KE4 Degeneration of DA neurons of nigrostriatal pathway |
AO Parkinsonian motor symptoms |
|
Rotenone 20-30 nM rat brain concentration [1-2] |
Approx. 53%[4-5] |
Approx. 20-53% (decrease in respiration rate)[1-2] |
Approx. 20-60% (decrease in UPS (26S) activity) [3] |
Neuronal loss (50% of animal affected) [2] |
Motor impairment (100% of animals with neuronal loss) [2] |
|
MPP+ 12-47 µM rat brain concentration [4-5] |
Approx. 50-75% [5] |
Approx. 38% (reduction in phosphorylating respiration) [5] |
Approx. 60% (decrease in UPS activity) [4] |
Approx. 50% of neuronal loss [4-5] |
Motor impairment [4] |
[1]; Okun et al., 1999 [2]; Barrientos and Moraes 1999; [3] Borland et al., 2008 [4] Thomas et al., 2012; [5] Betarbet et al., 2000 and 2006.
Summary of the proposed Key Events in this AOP:

Chronic, low level of exposure to environmental chemicals that inhibit complex I could result in mitochondrial dysfunction and oxidative stress, triggering proteasomal dysfunction strongly implicated in parkinsonian disorders, including aggregation/modifications in α-synuclein protein and organelles trafficking. These cellular key events cause DA terminals degeneration in striatum and progressive cell death of DA neurons in SNpc, accompanied by neuroinflammation that potentiates neuronal cell death, finally leading to parkinsonian's motor symptoms. Important to notice that at each step, the effects become regionally restricted such that systemic complex I inhibition eventually results in highly selective degeneration of the nigrostriatal pathway.
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
A preliminary quantitative AOP has been published in Tebby et al. (2022). This qAOP was developed by modelling the KER using a set of mathematical functions, for two chemicals, rotenone and deguelin, based on data obtained in LUHMES cells.
Complex I activity was measured using in proliferating LUHMES cells. Decrease in mitochondrial respiration was and measured using an Agilent® Seahorse OCR equipment. Mitochondrial respiration and proteasomal activity were measured using the same cells at a stage of neurite growth (day 3 of differentiation). The proteasomal function of cells was assessed at 24 h after toxicant exposure by a fluorogenic substrate that increases in fluorescence when the proteasome is active (Delp et al., 2021). Neuronal degeneration was represented by neurite area which was measured at a stage of neurite growth (day 2 of differentiation). The neurite areas (which serves as indirect measurement of neuronal interconnectivity) of stained differentiating neurons, as well as cellular viability were measured simultaneously at 24 h after toxicant exposure using high content imaging.
Each KER was modelled a mathematical equation, either A) a 4-parameter log-logistic (Hill) function often used for dose-response modelling, B) a 2-parameter linear function which implies equal EC50 for the two adjacent KEs, or C) an increasing function which increases towards a horizontal asymptote, with an optional horizontal shift, implying a higher EC50 in the downstream KE. A ramification for KE3 was modelled: neurite area (KE4) was modelled as the product of two functions of mitochondrial respiration (KE2) and proteasomal activity (KE3) under the assumption that both KEs acted independently on it. The model parameters were estimated in a Bayesian statistical framework independently for rotenone and deguelin. The results are described in the open access paper https://doi.org/10.1016/j.tiv.2022.105345 and available with the model code on Zenodo 10.5281/zenodo.5549494
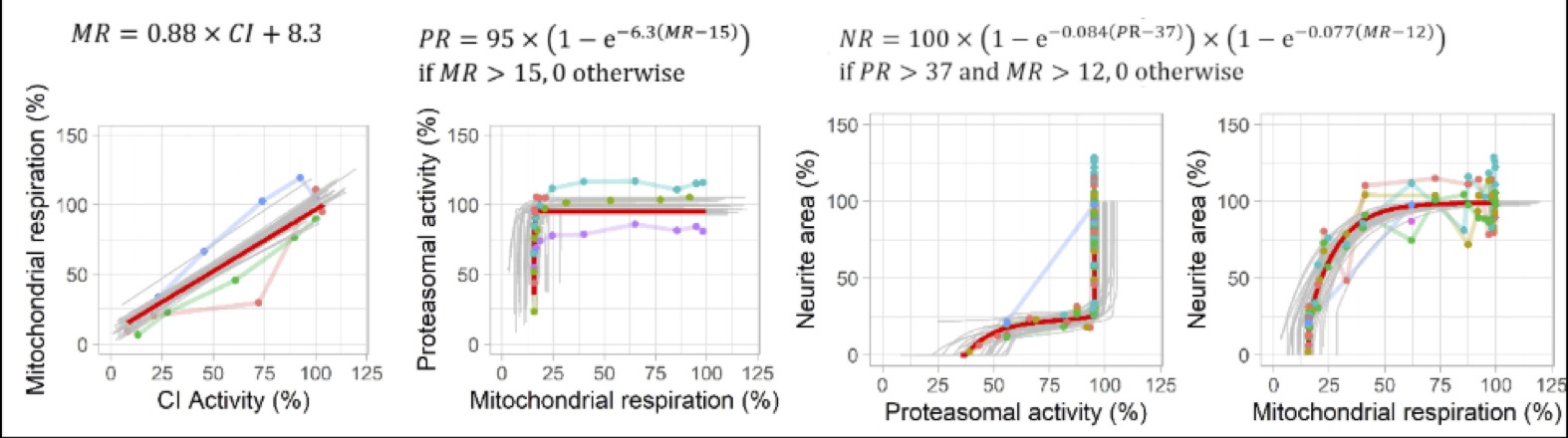
Figure 3: Predicted and observed KERs for rotenone for each readout (complex I activity, mitochondrial respiration, proteasomal activity, neurite area. Red line: predictions obtained with the maximum posterior vector. Grey lines: predictions obtained with 30 random parameter vectors drawn from their joint posterior distribution. Dots: observations of KEx at predicted KEx-1, colours represent replicates. (Reproduced from Tebby et al. 2022)
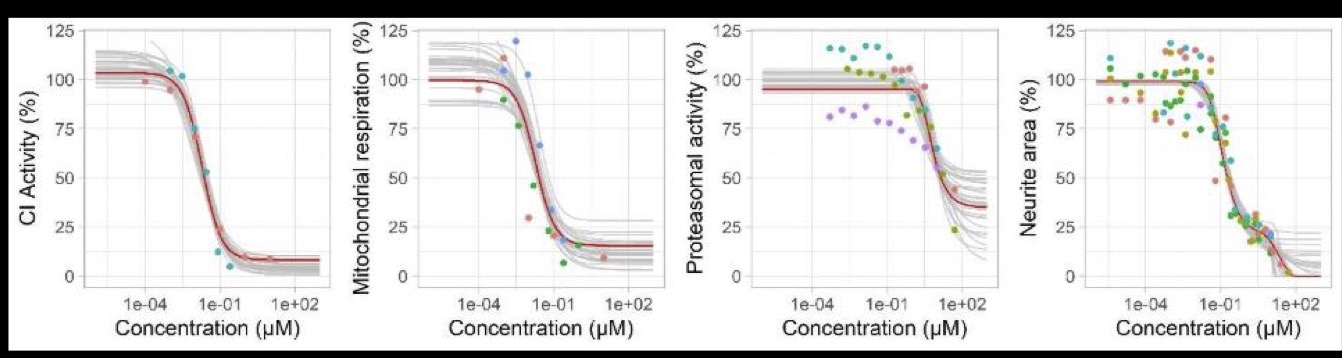
Figure 4: Predicted and observed dose-response relationships for rotenone for each readout (complex I activity, mitochondrial respiration, proteasomal activity, neurite area. Red line: predictions obtained with the maximum posterior parameter values. Grey lines: predictions obtained with 30 random parameter vectors drawn from their joint posterior distribution. Dots: experimental data, colours represent replicates. (Reproduced from Tebby et al. 2022)
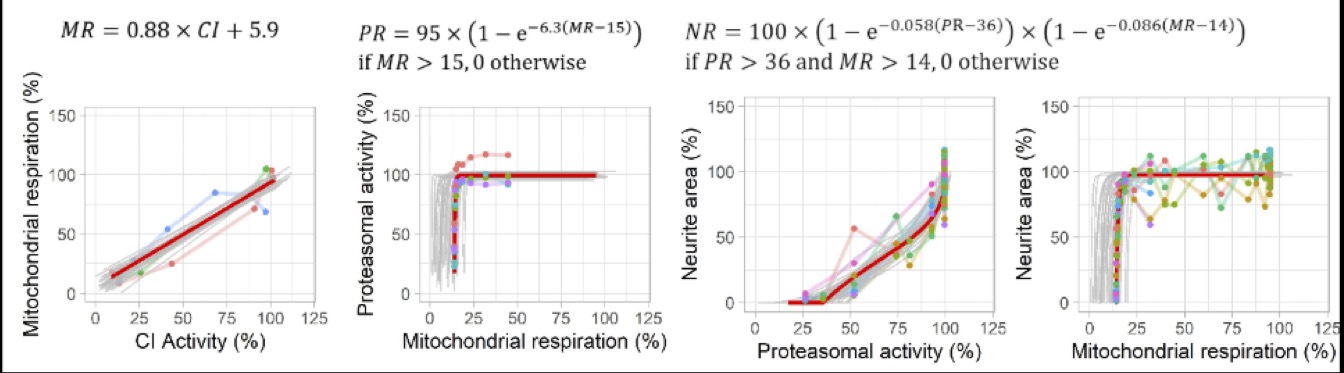
Figure 5: Predicted and observed KERs for deguelin for each readout (complex I activity, mitochondrial respiration, proteasomal activity, neurite area. Red line: predictions obtained with the maximum posterior vector. Grey lines: predictions obtained with 30 random parameter vectors drawn from their joint posterior distribution. Dots: observations of KEx at predicted KEx-1, colours represent replicates. (Reproduced from Tebby et al. 2022)
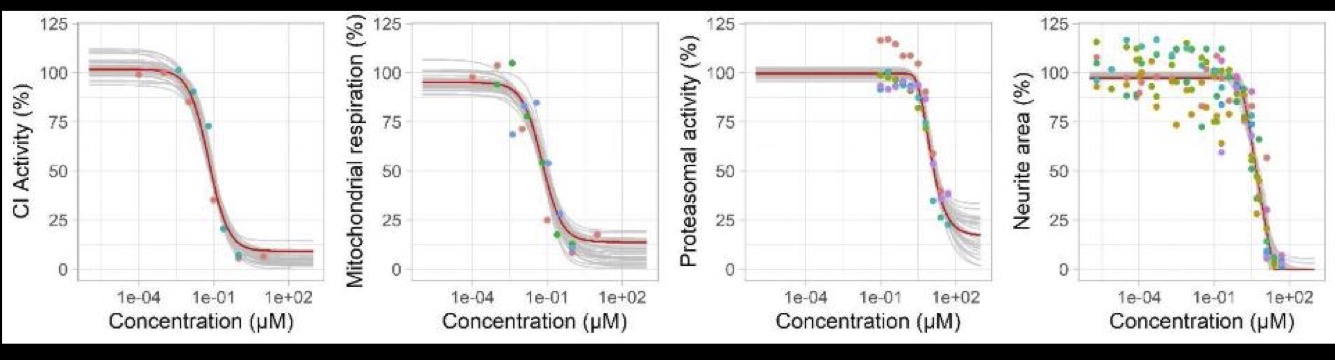
Figure 6: Predicted and observed dose-response relationships for deguelin for each readout (complex I activity, mitochondrial respiration, proteasomal activity, neurite area. Red line: predictions obtained with the maximum posterior parameter values. Grey lines: predictions obtained with 30 random parameter vectors drawn from their joint posterior distribution. Dots: experimental data, colours represent replicates. (Reproduced from Tebby et al. 2022)
Limitations and uncertainties
This preliminary qAOP, based on a limited set of data, can be extended to include data identified in the literature and listed in section empirical evidence for cI inhibitors KER 934 and KER 908 (part of Revision of AOP3 Project: NP/EFSA/PREV/2024/02), including for other stressors.
Data used to extend quantitative understanding was collected for a set of three compounds, rotenone, deguelin and tebufenpyrad, either by digitalizing summary data (mean and standard deviation) from publications, or by collecting data from the Biostudies online database. The data from Biostudies was visually compared to published data in order to establish correspondence between the datasets. Multiple-concentration data for cI inhibition (MIE/KE887) at 30 minutes was collected by digitalising figures from Alimohammadi et al. (2023) (tebufenpyrad) and from Biostudies S-TOX1365 (https://www.ebi.ac.uk/biostudies/eu-toxrisk/studies/S-TOXR1365) which could have been published in Tebby et al. (2022) (rotenone and deguelin). Multiple concentration data for OCR mitochondrial respiration (KE177) measured at 24 hours in LUHMES cells was collected from Alimohammadi et al. (2023) (tebufenpyrad) and from Biostudies S-TOXR1315 (https://www.ebi.ac.uk/biostudies/eu-toxrisk/studies/S-TOXR1315) which could have been published in Tebby et al. (2022) and van der Stel et al. (2020) (rotenone and deguelin). Multiple concentration data for ATP content (KE177) measured at 24 hours in LUHMES cells was collected from Biostudies S-TOXR1683 (https://www.ebi.ac.uk/biostudies/eu-toxrisk/studies/S-TOXR1683) which could have been published in Delp et al. (2021) (rotenone, deguelin, tebufenpyrad). Multiple concentration data for cell viability and neurite area (KE890) measured at 24 hours in LUHMES cells was collected from Biostudies S-TOXR1203 (https://www.ebi.ac.uk/biostudies/eu-toxrisk/studies/S-TOXR1203) which could have been published in Delp et al. (2021) (rotenone, deguelin, tebufenpyrad), from Biostudies S-TOXR1279 (https://www.ebi.ac.uk/biostudies/eu-toxrisk/studies/S-TOXR1279) which could have been published in van der Stel et al. (2020) (rotenone, deguelin) and from raw data provided by the authors of Alimohammadi et al. (2023) (tebufenpyrad).
Besides the uncertainty quantified by the statistical analysis including the individual replicate data, several sources of uncertainty are not accounted for in this preliminary qAOP.
Each KE can be represented by several readouts; reassessment of the qAOP from Tebby et al. (2022) highlighted the following limitations regarding the choice of endpoints representing the KEs and regarding the KER between cI inhibition and the first KE.
Neuron degeneration may not be adequately represented by neurite area measured in differentiating LUHMES cells during a 24-hour exposure starting a day 2 of differentiation. Repeated exposure in differentiated cells should be investigated. Furthermore, the KER between decrease in mitochondrial respiration and decrease in neurite area appeared to be substance specific when considering both rotenone and tebufenpyrad. A possible explanation is that effects of rotenone on microtubules could decrease neurite area at lower exposure concentrations than expected based on tebufenpyrad data and mitochondrial respiration data (recall the previously cited reference).
Cell viability could be a relevant alternative to neurite area for representing neuron degeneration, however, the quantitative assessment revealed that the decrease in cell viability occurred at exposure concentrations at which mitochondrial respiration was already at its lowest level. Since both endpoints were measured in the same type of assay at the same timepoint, the exposure concentration levels between both endpoints are comparable. The lack of overlap between decrease of mitochondrial respiration and of cell viability suggests that other mechanisms are involved between these two key events or that the 24-hour exposure timepoint is not the most relevant.
Several measurements of KE2 are possible: the decrease in ATP production could be an alternative endpoint to the decrease in mitochondrial respiration. Data on the resulting decrease in ATP content was available and was envisioned as a measurement of a downstream key event of mitochondrial respiration. However, decrease in mitochondrial respiration and in ATP content did not overlap in terms of effective concentration levels.
The lack of overlap in effective concentrations between decreases in mitochondrial respiration and ATP content or cell viability may be due to the ability of cells to switch from oxidative phosphorylation to glycolysis for energy production in the presence of glucose, compensating for mitochondrial inhibition. Assays using galactose as a substrate avoid this compensatory pathway. Nevertheless, dose-response data available in the literature had been mostly obtained in assays using glucose as a substrate rather than galactose. To enhance qAOP and lower KER uncertainty, more data in galactose conditions is needed to quantify the KERs using data obtained in LUHMES cells. Fortunately, the PANDORA project (OC/EFSA/PREV/2023/0, Environmental Neurotoxicants – Advancing Understanding on the Impact of Chemical Exposure on Brain Health and Disease, LOT 3) is currently producing such data.
Endpoints measured in the same experimental setup allow for comparisons of effective concentrations along the AOP. Complex I inhibition was measured in permeabilized cells, whereas the other endpoints that were quantitatively assessed were measured in intact cells. Intracellular exposure concentrations are therefore likely different and dependent on toxicokinetics. Chemical agnosticity of the KER between complex I inhibition and the decrease in mitochondrial respiration cannot be expected. The available data showed similar effective concentrations, but since the concentration levels in both assays may not be directly comparable due to differences in toxicokinetics, lack of overlap would not suggest a missing intermediate key event. Without intracellular concentration data, the comparison of effective concentrations in dissimilar assays provides only limited quantitative understanding of the KER.
When exposure concentrations between adjacent KEs are comparable, quantitative AOP modelling would benefit from application of FAIR principles to data. Publication of raw data with metadata that could indicate which data were obtained with identical biological material and experimental conditions would help refine quantitative KERs. Data rich compounds such as rotenone sometimes showed large variations in potencies for a given endpoint.
Overall, reassessment of the available data considering the preliminary qAOP developed by Tebby et al. (2022) highlighted several limitations inherent to the data used for qAOPs, in particular comparability of exposure concentrations between the various endpoints, comparability and relevance of timepoints and exposure durations, the necessary overlap in effective concentrations between adjacent endpoints that are measured at comparable exposure concentrations and relevant timepoints, and data reproducibility. Selection of most relevant readouts and accurate characterization of the molecular initiating event for cross-validation are critical when designing in vitro experiments targeted at calibrating qAOPs.
According to the limits that were discussed, the following recommendations are made:
As cultured cells rely predominantly on glycolysis as their primary source of ATP, it is recommended to replace glucose with galactose to shift cellular energy metabolism towards mitochondrial oxidative phosphorylation and increase cellular dependence on mitochondrial respiration.
Consider a shorter exposure time of less than 24 hours for KE177 and increasing exposure duration over the 24h to identify potential effects on KE890
Consider using differentiated LUHMES cells rather than DoD2 to DoD3 to avoid the contribution of developmental pathways (e.g. neurite outgrowth) not any more active in a mature state.
Future developments of qAOP should include methods for modelling non overlapping adjacent Kes and research on the development and application of biokinetic models for in vitro data that would accurately predict the internal concentration causing the effects.
- Not endorsed
Considerations for Potential Applications of the AOP (optional)
- This AOP has been developed in order to evaluate the biological plausibility that the adverse outcome i.e. parkinsonian motor deficits, is linked to a MIE that can be triggered by chemical substances i.e. pesticides and chemicals in general. The relevance of the AOP has been documented by tools compounds known to trigger the described AOP. By means of using a human health outcome that has been shown in epidemiological studies to be association with pesticide exposure, the authors intend to draw attention on this AO in the process of hazard identification. This AOP can be used to support the biological plausibility of this association during the process of evaluation and integration of the epidemiological studies into the risk assessment. It is biologically plausible that a substance triggering the pathway, can be associated with the AO and ultimately with the human health outcome, pending the MoA analysis.
- In addition, this AOP can be used to support identification of data gaps that should be explored when a chemical substance is affecting the pathway. Moreover, the AOP provides a scaffold for recommendations on the most adequate study design to investigate the apical endpoints. It is important to note that, although the AO is defined in this AOP as parkinsonian motor deficits, degeneration of DA neurons is already per se an adverse outcome even in situations where it is not leading to parkinsonian motor deficits, and this should be taken into consideration for the regulatory applications of this AOP.
- The MIE and KEs identified in this AOP could serve as a basis for assays development that could contribute to an AOP informed-IATA construction which can be applied for different purposes such as: screening and prioritization of chemicals for further testing, hazard characterization or even risk assessment when combined with exposure and ADME information.
-
This AOP can be used for neurotoxicity assessment, since it is plausible that a compound that binds to the mitochondrial complex I may eventually lead to Parkinsonian motor deficits.
-
The regulatory applicability of this AOP would be to use experimental findings in model systems representing the MIE and KEs as indicators/alerts for the AO. Risk assessment may be possible if bioavailability at the target cells can be estimated, the toxic concentrations in vitro can be extrapolated to in vivo and exposure scenarios can be simulated.
-
This AOP can be applied for chemicals that have structural similarities to rotenone or MPTP. However, this AOP may not at the moment be used for chemicals that do not resemble rotenone or MPTP. It is however expected that compounds acting on the same MIE, but belonging to different chemical classes and those that are structurally different, will be tested in the near future in order to substantiate a broader specificity for this AOP. However, it remains evident that chemicals affecting the MIE are potential risk factors for this AO.
References
Alimohammadi Mahshid, Birthe Meyburg, Anna-Katharina Ückert, Anna-Katharina Holzer, Marcel Leist, 2023. EFSA Pilot Project on New Approach Methodologies (NAMs) for Tebufenpyrad Risk Assessment. Part 2. Hazard characterisation and identification of the Reference Point. EFSA supporting publication 2023:EN-7794. 56 pp. doi:10.2903/sp.efsa.2023.EN-7794
Abdelsalam RM, Safar MM J Neurochem. Neuroprotective effects of vildagliptin in rat rotenone Parkinson's disease model: role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. 2015 Jun;133(5):700-7. doi: 10.1111/jnc.13087. Epub 2015 Mar 26.
Aguilar JS, Kostrzewa RM. Neurotoxin mechanisms and processes relevant to parkinson’s disease: un update. Neurotox Res. DOI 10.1007/s12640-015-9519-y.
Alvarez-Fischer D, Guerreiro S, Hunot S, Saurini F, Marien M, Sokoloff P, Hirsch EC, Hartmann A, Michel PP. Modelling Parkinson-like neurodegeneration via osmotic minipump delivery of MPTP and probenecid. J Neurochem. 2008 Nov;107(3):701-11. doi: 10.1111/j.1471-4159.2008.05651.x. Epub 2008 Sep 16.
Arnold, B., et al. (2011). "Integrating Multiple Aspects of Mitochondrial Dynamics in Neurons: Age-Related Differences and Dynamic Changes in a Chronic Rotenone Model." Neurobiology of Disease 41(1): 189-200.
Barbeito AG, Mesci P, Boillee S. 2010. Motor neuron-immune interactions: the vicious circle of ALS. J Neural Transm 117(8): 981-1000.
Balmaceda V, Komlódi T, Szibor M, Gnaiger E, Moore AL, Fernandez-Vizarra E, Viscomi C. The striking differences in the bioenergetics of brain and liver mitochondria are enhanced in mitochondrial disease. Biochim Biophys Acta Mol Basis Dis. 2024 Mar;1870(3):167033. doi: 10.1016/j.bbadis.2024.167033. Epub 2024 Jan 26. PMID: 38280294.
Barrientos A., and Moraes C.T. (1999) Titrating the Effects of Mitochondrial Complex I Impairment in the Cell Physiology. Vol. 274, No. 23, pp. 16188–16197.
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973 Dec;20(4):415-55
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 3:1301–6
Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG,Mc Lendon C, Kim JH, Lund S, Na HM, taylor G, Bence NF, kopito R, seo BB, Yagi T, Yagi A, Klinfelter G, Cookson MR, Greenmyre JT. 2006. Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, α-synuclein, and the ubiquitin-proteasome system. Neurobiology disease. (22) 404-20.
Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie JM, Gross CE (2001) Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson's disease. J Neurosci. 21(17):6853-61.
Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B. 2004. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging cell 3(4): 169-176.
Blesa J, Pifl C, Sánchez-González MA, Juri C, García-Cabezas MA, Adánez R, Iglesias E, Collantes M, Peñuelas I, Sánchez-Hernández JJ, Rodríguez-Oroz MC, Avendaño C, Hornykiewicz O, Cavada C, Obeso JA (2012) The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: a PET, histological and biochemical study. Neurobiol Dis. 48(1):79-91.
Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, et al. 2014. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci 34(25): 8546-8556.
Borrajo A, Rodriguez-Perez AI, Villar-Cheda B, Guerra MJ, Labandeira-Garcia JL. 2014. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology 85: 1-8
Brinkley BR, Barham SS, Barranco SC, and Fuller GM. 1974. Rotenone inhibition of spindle microtubule assembly in mammalian cells,” Experimental Cell Research. 85(1)41–46.
Brown GC, Bal-Price A (2003) Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27: 325-355
Braun RJ. (2012). Mitochondrion-mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Front Oncol 2:182.
Brzozowski MJ, Jenner P, Rose S. 2015. Inhibition of i-NOS but not n-NOS protects rat primary cell cultures against MPP(+)-induced neuronal toxicity. J Neural Transm 122(6): 779-788.
Burr SP, Klimm F, Glynos A, Prater M, Sendon P, Nash P, Powell CA, Simard ML, Bonekamp NA, Charl J, Diaz H, Bozhilova LV, Nie Y, Zhang H, Frison M, Falkenberg M, Jones N, Minczuk M, Stewart JB, Chinnery PF. Cell lineage-specific mitochondrial resilience during mammalian organogenesis. Cell. 2023 Mar 16;186(6):1212-1229.e21. doi: 10.1016/j.cell.2023.01.034. Epub 2023 Feb 23. PMID: 36827974.
Cacquevel M, Lebeurrier N, Cheenne S, Vivien D. 2004. Cytokines in neuroinflammation and Alzheimer's disease. Curr Drug Targets 5(6): 529-534.
Calne DB, Sandler M (1970) L-Dopa and Parkinsonism. Nature. 226(5240):21-4.
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT (2009) A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis. 34(2):279-90.
Cappelletti G, Maggioni MG, Maci R. 1999. Influence of MPP+ on the state of tubulin polymerisation in NGF-differentiated PC12 cells. J Neurosci Res. 56(1):28-35.
Cao S, Theodore S, Standaert DG.2010. Fc gamma receprors are required for NF-kB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson's disease. molecular neurodegeneration.5-42.
Cappelletti G, Pedrotti B, Maggioni MG, Maci R. 2001. Microtubule assembly is directly affected by MPP(+)in vitro. Cell Biol Int.25(10):981-4.
Castrioto A, Lozano AM, Poon YY, Lang AE, Fallis M, Moro E. 2011. Ten-year outcome of subthalamic stimulation in Parkinson disease: a blinded evaluation. Arch Neurol. 68(12):1550-6.
Chang CY, Choi DK, Lee DK, Hong YJ, Park EJ. 2013. Resveratrol confers protection against rotenone-induced neurotoxicity by modulating myeloperoxidase levels in glial cells. PLoS One 8(4): e60654.
Champy et al. (2004). Annonacin, a lipophilic inhibitor of mitochondrial complex I, induces nigral and striatal neurodegeneration in rats: possible relevance for atypical Parkinsonism in Guadeloupe. J Neurochem 88: 63-69.
Chao YX, He BP, Tay SS. 2009. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson's disease. J Neuroimmunol 216(1-2): 39-50.
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF, Xu JP 2015. Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Mol Neurodegener. 2;10(1):4.
Chinta SJ, Andersen JK (2006) Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radic Biol Med. 41(9):1442-8.
Choi WS., Kruse S.E., Palmiter R, Xia Z., (2008) Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP, or paraquat. PNAS, 105, 39, 15136-15141
Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ (2014) Celastrol from 'Thunder God Vine' protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson's disease. Neurochem Res. 39(1):84-96.
Chiu CC, Yeh TH, Lai SC, Wu-Chou YH, Chen CH, Mochly-Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS. 2015. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp Neurol. 263:244-53.
Chou AP, Li S, Fitzmaurice AG, Bronstein JM. 2010. Mechanisms of rotenone-induced proteasome inhibition. NeuroToxicology. 31:367–372. Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, et al. 2011. Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology 60(6): 963-974.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. (2012). Mitochondrial importance in Alzheimer’s, Huntington’s and Parkinson’s diseases. Adv Exp Med Biol 724:205 – 221.
Cotzias GC, Papavasiliou PS, Gellene R. 1969. L-dopa in parkinson's syndrome. N Engl J Med. 281(5):272.
Cozzolino M, Ferri A, Valle C, Carri MT. (2013). Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci 55:44 – 49.
Dagda RK, Banerjee TD and Janda E. 2013. How Parkinsonian Toxins Dysregulate the Autophagy Machinery. Int. J. Mol. Sci. 14:22163-22189.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. 2002. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 99(22):14524-9.
Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. 2002. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 99(22):14524-9.
Dauer W, Przerdborski S. 2003. Parkinson’sdisease: Mechanisms and Models.Neuron. 39, 889-9.
De Bie RM, de Haan RJ, Nijssen PC, Rutgers AW, Beute GN, Bosch DA, Haaxma R, Schmand B, Schuurman PR, Staal MJ, Speelman JD. 1999. Unilateral pallidotomy in Parkinson's disease: a randomised, single-blind, multicentre trial. Lancet. 354(9191):1665-9.
Degli Esposti M, Ghelli A. 1994. The mechanism of proton and electron transport in mitochondrial complex I. Biochim Biophys Acta.1187(2):116–120.
Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. 2010. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30:12535–12544.
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. 2000. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem 74(5): 2213-2216.
Dehmelt, L and Shelley H. Neurite Outgrowth: A Flick of the Wrist. Current Biology, 2007 Volume 17, Issue 15, R611 – R614
Delp J, Cediel-Ulloa A, Suciu I, Kranaster P, van Vugt-Lussenburg BM, Munic Kos V, van der Stel W, Carta G, Bennekou SH, Jennings P, van de Water B, Forsby A, Leist M. Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors. Arch Toxicol. 2021 Feb;95(2):591-615. doi: 10.1007/s00204-020-02970-5. Epub 2021 Jan 29. PMID: 33512557; PMCID: PMC7870626.
Delp J, Funke M, Rudolf F, Cediel A, Bennekou SH, van der Stel W, Carta G, Jennings P, Toma C, Gardner I, van de Water B, Forsby A, Leist M. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch Toxicol. 2019 Jun;93(6):1585-1608. doi: 10.1007/s00204-019-02473-y. Epub 2019 Jun 12. PMID: 31190196.
Deuschl G, Schade-Brittinger C, Krack P, Volkmann J, Schäfer H, Bötzel K, Daniels C, Deutschländer A, Dillmann U, Eisner W, Gruber D, Hamel W, Herzog J, Hilker R, Klebe S, Kloss M, Koy J, Krause M, Kupsch A, Lorenz D, Lorenzl S, Mehdorn HM, Moringlane JR, Oertel W, Pinsker MO, Reichmann H, Reuss A, Schneider GH, Schnitzler A, Steude U, Sturm V, Timmermann L, Tronnier V, Trottenberg T, Wojtecki L, Wolf E, Poewe W, Voges J; German Parkinson Study Group, Neurostimulation Section. 2006. A randomized trial of deep-brain stimulation for Parkinson's disease. N Engl J Med. 355(9):896-908.
Dexter D. T., Jenner P.. Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biology and Medicine 62 (2013) 132-144
Dietz GPH, Stockhausen KV, Dietz B et al. (2008) Membrane-permeable Bcl-xL prevents MPTP-induced dopaminergic neuronal loss in the substantia nigra. J Neurochem 104:757-765. Doi:10.1111/j.1471-4159.2007.05028.
Drolet RE, Behrouz B, Lookingland KJ, Goudreau JL, 2004. Mice lacking α-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology. 25(5):761-9.
Drouin-Ouellet J, St-Amour I, Saint-Pierre M, Lamontagne-Prolux J, Kriz J, Barker R, Cicchetti F.2015. Toll-like receptor expression in the blood and brain of patients and a mouse of Parkinson's disease. International Journal of Neuropsychopharmacology. 1-11.
Du T, Li L, Song N, Xie J, Jiang H (2010) Rosmarinic acid antagonized 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in MES23.5 dopaminergic cells. Int J Toxicol. 29(6):625-33.
Efremov RG, Sazanov LA. Respiratory complex I: 'steam engine' of the cell? Curr Opin Struct Biol. 2011 Aug;21(4):532-40. doi: 10.1016/j.sbi.2011.07.002. Epub 2011 Aug 8. Review.
Efremov RG, Sazanov LA. Structure of the membrane domain of respiratory complex I. Nature. 2011 Aug 7;476(7361):414-20. doi: 10.1038/nature10330.
EFSA Panel on Plant Protection Products and their residues (PPR); Ockleford C, Adriaanse P, Berny P, Brock T, Duquesne S, Grilli S, Hernandez-Jerez AF, Bennekou SH, Klein M, Kuhl T, Laskowski R, Machera K, Pelkonen O, Pieper S, Smith R, Stemmer M, Sundh I, Teodorovic I, Tiktak A, Topping CJ, Wolterink G, Angeli K, Fritsche E, Hernandez-Jerez AF, Leist M, Mantovani A, Menendez P, Pelkonen O, Price A, Viviani B, Chiusolo A, Ruffo F, Terron A, Bennekou SH. Investigation into experimental toxicological properties of plant protection products having a potential link to Parkinson's disease and childhood leukaemia. EFSA J. 2017 Mar 16;15(3):e04691. doi: 10.2903/j.efsa.2017.4691. PMID: 32625422; PMCID: PMC7233269.
Emmrich JV, Hornik TC, Neher JJ, Brown GC. 2013. Rotenone induces neuronal death by microglial phagocytosis of neurons. The FEBS journal 280(20): 5030-5038.
Esposti et al. (1993) Complex I and Complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem Biophys Res Commun 190(3): 1090-6.
Fasano A, Romito LM, Daniele A, Piano C, Zinno M, Bentivoglio AR, Albanese A. 2010. Motor and cognitive outcome in patients with Parkinson's disease 8 years after subthalamic implants. Brain. 133(9):2664-76.
Fato et al. (2009) Differential effects of mitochondrial complex I inhibitors on production of reactive oxygen species. Biochim Biophys Acta 1787(5): 384-392.
Feng ZH, Wang TG, Li DD, Fung P, Wilson BC, Liu B, et al. 2002. Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1, 2, 3, 6-tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci Lett 329(3): 354-358.
Feng J. Mictrotubule. A common target for parkin and Parkinson's disease toxins. Neuroscientist 2006, 12.469-76.
Ferger B, Leng A, Mura A, Hengerer B, Feldon J. 2004. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem 89(4): 822-833.
Ferrante RJ, Schulz JB, Kowall NW, Beal MF. 1997. Systematic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain Research. (753). 157-2.
Ferrari-Toninelli G, Bonini SA, Cenini G, Maccarinelli G, Grilli M, Uberti D, Memo M. 2008. Dopamine receptor agonists for protection and repair in Parkinson's disease. Curr Top Med Chem. 8(12):1089-99.
Friedman LG, lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. 2012. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. The Journal of Neuroscience. 32 (22) 7585-93.
Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti C, Ruffoli R, Soldani P, Ruggieri S, Alessandri’ MG, Paparelli A. 2003. Fine structure and mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. The journal of neuroscience. 23 (26) 8955-6.
Fornai F., P. Lenzi, M. Gesi et al., “Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells,” Journal of Neurochemistry, vol. 88, no. 1, pp. 114–123, 2004.
Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Südhof TC. 2005. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitinproteasome system and _α-synuclein. PNAS. 102: 3413–3418.
Freed CR, Breeze RE, Rosenberg NL, Schneck SA, Wells TH, Barrett JN, Grafton ST, Huang SC, Eidelberg D, Rottenberg DA. 1990.
Transplantation of human fetal dopamine cells for Parkinson's disease. Results at 1 year. Arch Neurol. 47(5):505-12.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP, Eifes S, Del Sol A, Schneider R, Kitano H, Balling R. 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Mol Neurobiol.49(1):88-102.
Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Molecular Cell. 2009;33:627–638.
Gao HM, Hong JS, Zhang W, Liu B. 2002. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22(3): 782-790.
Gao HM, Liu B, Hong JS. 2003. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 23(15): 6181-6187.
Gao L, Brenner D, Llorens-Bobadilla E, Saiz-Castro G, Frank T, Wieghofer P, et al. 2015. Infiltration of circulating myeloid cells through CD95L contributes to neurodegeneration in mice. J Exp Med 212(4): 469-480.
González-Rodríguez, P., Zampese, E., Stout, K.A. et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599, 650–656 (2021). https://doi.org/10.1038/s41586-021-04059-0
Graier WF, Frieden M, Malli R. (2007). Mitochondria and Ca2+ signaling: old guests, new functions. Pflugers Arch 455:375–396.
Greenamyre, J T., Sherer, T.B., Betarbet, R., and Panov A.V. (2001) Critical Review Complex I and Parkinson’s Disease Life, 52: 135–141.
Greenamayre et al. 2010. Lessons from the rotenone model of Parkinson's disease. Trends pharmacol. Sci. 31(4):141-2
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, et al. 1998. Glial-neuronal interactions in Alzheimer's disease: the potential role of a 'cytokine cycle' in disease progression. Brain Pathol 8(1): 65-72.
Grivennikova, V.G., Maklashina, E.O., E.V. Gavrikova, A.D. Vinogradov (1997) Interaction of the mitochondrial NADH-ubiquinone reductase with rotenone as related to the enzyme active/inactive transition Biochim. Biophys. Acta, 1319 (1997), pp. 223–232
Gusdon AM, Fernandez-Bueno GA, Wohlgemuth S, Fernandez J, Chen J, Mathews CE. Respiration and substrate transport rates as well as reactive oxygen species production distinguish mitochondria from brain and liver. BMC Biochem. 2015 Sep 10;16:22. doi: 10.1186/s12858-015-0051-8. PMID: 26358560; PMCID: PMC4564979.
Hajieva P, Mocko JB, Moosmann B, Behl C (2009) Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. J Neurochem. 110(1):118-32.
Hoglinger G.U. et al.2003.Chronic systemic complex I inhibition induces a hypokynetic multisystem degeneration in rats. J.neurochem 84:491-502.
Hornykiewicz O, Kish SJ. 1987. Biochemical pathophysiology of parkinson’s disease. In Parkinson’s Disease. M Yahr and K.J. Bergmann, eds (New.York: Raven Press) 19-34.
Jana S, Sinha M, Chanda D, Roy T, Banerjee K, Munshi S, Patro BS, Chakrabarti S (2011) Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson's disease. Biochim Biophys Acta. 1812(6):663-73.
Jha N, Jurma O, Lalli G, Liu Y, Pettus EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK (2000) Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. J Biol Chem. 275(34):26096-101.
Johnson ME, Bobrovskaya L. 2015. An update on the rotenone models of parkinson’s disease: Their ability to reproduce features of clinical disease and model gene-environment interactions. 946). 101-16.
Heikkila RE, Nicklas WJ, Vyas I, Duvoisin RC. 1985. Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci Lett. 62(3):389-94.
Henderson BT, Clough CG, Hughes RC, Hitchcock ER, Kenny BG. 1991. Implantation of human fetal ventral mesencephalon to the right caudate nucleus in advanced Parkinson's disease. Arch Neurol. 48(8):822-7.
Hirsch EC, Hunot S. 2009. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8(4): 382-397. Hoglinger GU, Feger J, Annick P, Michel PP, Karine P, Champy P, Ruberg M, Wolfgang WO, Hirsch E. 2003. Chronic systemic complexI inhibition induces a hypokinetic multisystem degeneration in rats. J. Neurochem.. (84) 1-12.
Ichimaru N, Murai M, Kakutani N, Kako J, Ishihara A, Nakagawa Y, Miyoshi H. 2008.. Synthesis and Characterization of New Piperazine-Type Inhibitors for Mitochondrial NADH-Ubiquinone Oxidoreductase (Complex I). Biochemistry. 47(40)10816–10826.
Inden M, Yoshihisa Kitamura, Hiroki Takeuchi, Takashi Yanagida, Kazuyuki Takata, Yuka Kobayashi, Takashi Taniguchi, Kanji Yoshimoto, Masahiko Kaneko, Yasunobu Okuma, Takahiro Taira, Hiroyoshi Ariga and Shun Shimohama. 2007. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. Journal of Neurochemistry. 101.(6).1491–4.
Keeney PM,Xie J,Capaldi RA,Bennett JP Jr. (2006) Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 10;26(19):5256-64.
Kelly PJ, Ahlskog JE, Goerss SJ, Daube JR, Duffy JR, Kall BA. 1987. Computer-assisted stereotactic ventralis lateralis thalamotomy with microelectrode recording control in patients with Parkinson's disease. Mayo Clin Proc. 62(8):655-64.
Khan MM, Kempuraj D, Zaheer S, Zaheer A. 2014. Glia maturation factor deficiency suppresses 1-methyl-4-phenylpyridinium-induced oxidative stress in astrocytes. J Mol Neurosci 53(4): 590-599.
Kim-Han JS, Dorsey JA, O’Malley KL. 2011. The parkinsonian mimetic MPP+, specifically impairs mitochondrial transport in dopamine axons. The Journal of Neuroscience. 31(19) 7212-1.
Kirk D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel R, Bijorklund A. 2002. Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. 22(7) 2780-91.
Kirk D, Annett L, Burger C, Muzyczka N, Mandel R, Bijorklund A. 2003. Nigrostriatal α-synucleinopathy induced by viral vector-mediated overexpression of human α-synuclein: A new primate model of parkinson’s disease. PNAS (100) 2884-9.
Klein RL, King MA, Hamby ME, Meyer EM. 2002. Dopaminergic cell loss induced by human A30P α-synuclein gene transfer to the rat substantia nigra. Hum.Gene.Ther. (13) 605-2.
Koller WC (1992) When does Parkinson's disease begin? Neurology. 42(4 Suppl 4):27-31 Koopman W, Hink M, Verkaart S, Visch H, Smeitink J, Willems P. 2007. Partial complex I inhibition decreases mitochondrial motility and increases matrix protein diffusion as revealed by fluorescence correlation spectroscopy. Biochimica et Biophysica Acta 1767:940-947.
Koopman W, Willems P (2012) Monogenic mitochondrial disorders. New Engl J Med. 22;366(12):1132-41. doi: 10.1056/NEJMra1012478.
Kraft AD, Harry GJ. 2011. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International journal of environmental research and public health 8(7): 2980-3018.
Lagoa et al. (2011) Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochimica et Biophys Acta 1807: 1562-1572.
Lang AE, Lozano AM. 1998. Parkinson's disease. Second of two parts. N Engl J Med. 339(16):1130-43.
Langston JW, ballard P, Irwin I. 1983. Chronic parkinsonism in human due to a product of meperidine-analog synthesis. Science. (219) 979-0.
Lapointe N, StHilaire M, martinoli MG, Blanchet J, gould P, Rouillard C, Cicchetti F. 2004. Rotenone induces non-specific central nervous system and systemic toxicity. The FASEB Journal express article 10.1096/fj.03-0677fje
Lauwers E, Debyser Z, Van Drope J, DeStrooper B, Nuttin B. 2003. Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector-mediated overexpression of α-synuclein. Brain pathol. (13) 364-72.
Lesner, N. P., Wang, X., Chen, Z., Frank, A., Menezes, C. J., House, S., ... & Mishra, P. (2022). Differential requirements for mitochondrial electron transport chain components in the adult murine liver. Elife, 11, e80919.
Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, et al. 2012. alpha7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation 9: 98.
Liu Y, Li W, Tan C, Liu X, Wang X, Gui Y, Qin L, Deng F, Hu C, Chen L. 2014. Meta-analysis comparing deep brain stimulation of the globus pallidus and subthalamic nucleus to treat advanced Parkinson disease. J Neurosurg. 121(3):709-18.
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, et al. 2015. Activation of alpha7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson's disease. Neuropharmacology 91: 87-96.
Liu W, Kong S, Xie Q, Su J, Li W, Guo H, Li S, Feng X, Su Z, Xu Y, Lai X. Protective effects of apigenin against 1-methyl-4-phenylpyridinium ion induced neurotoxicity in PC12 cells. Int J Mol Med. 2015, 35(3):739-46.
Lloyd KG, Davidson L, Hornykiewicz O (1975) The neurochemistry of Parkinson's disease: effect of L-dopa therapy. J Pharmacol Exp Ther. 195(3):453-64.
Lo Bianco C, Ridet JL, Deglon N, Aebischer P. 2002. Alpha-synucleopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc.natl.Sci.USA (99)10813-8.
López-Lozano JJ, Bravo G, Abascal J. 1991. Grafting of perfused adrenal medullary tissue into the caudate nucleus of patients with Parkinson's disease. Clinica Puerta de Hierro Neural Transplantation Group. J Neurosurg. 75(2):234-43.
Mangano EN, Litteljohn D, So R, Nelson E, Peters S, Bethune C, et al. 2012. Interferon-gamma plays a role in paraquat-induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiol Aging 33(7): 1411-1426.
Marella M, Seo BB, Nakamaru-Ogiso E, Greenamyre JT, Matsuno-Yagi A, Yagi T (2008) Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. PLoS One. 3(1):e1433.
Marshall, L. E. & Himes, R. H. Rotenone inhibition of tubulin self-assembly. Biochim Biophys Acta 543, 590–594 (1978).
Martin LJ. (2011). Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43:569 – 579.
Matsumoto K, Asano T, Baba T, Miyamoto T, Ohmoto T. 1976. Long-term follow-up results of bilateral thalamotomy for parkinsonism. Appl Neurophysiol. 39(3-4):257-60.
McGeer PL, McGeer EG. 1998. Glial cell reactions in neurodegenerative diseases: Pathophysiology and therapeutic interventions. Alzheimer DisAssocDisord 12 Suppl. 2: S1-S6.
McNaught KS, Jenner P. 2001. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 297, 191– 194. McNaught KSC, Olanow W, Halliwell B. 2001. Failure of the ubiquitine-proteasome system in parkinson’s disease. Nature Rev. Neurosci. (2) 589-4.
McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. 2003. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 179, 38– 46.
Moldovan AS, Groiss SJ, Elben S, Südmeyer M, Schnitzler A, Wojtecki L. 2015. The treatment of Parkinson's disease with deep brain stimulation: current issues. Neural Regen Res. 10(7):1018-22.
Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D. (2012) Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci. 69(7):1153-65.
Narabayashi H, Yokochi F, Nakajima Y. 1984. Levodopa-induced dyskinesia and thalamotomy. J Neurol Neurosurg Psychiatry. 47(8):831-9. Nataraj J, Manivasagam T, Justin Thenmozhi A, Essa MM 2015. Lutein protects dopaminergic neurons against MPTP-induced apoptotic death and motor dysfunction by ameliorating mitochondrial disruption and oxidative stress. Nutr Neurosci. 2015 Mar 2. [Epub ahead of print].
Obeso JA, Rodríguez-Oroz MC, Rodríguez M, Lanciego JL, Artieda J, Gonzalo N, Olanow CW (2000) Pathophysiology of the basal ganglia in Parkinson's disease. Trends Neurosci. 23(10 Suppl):S8-19.
Offen D, Beart PM, Cheung NS et al. (1998) Transegnic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine neurotoxicity. PNAS 95:5789-5794
O’Malley KL. 2010. The role of axonopathy in Parkinson’s disease. 2010. Experimental Neurobiology. (19). 115-19.
Okun, J.G, Lümmen, P and Brandt U., (1999) Three Classes of Inhibitors Share a Common Binding Domain in Mitochondrial Complex I (NADH:Ubiquinone Oxidoreductase) J. Biol. Chem. 274: 2625-2630. doi:10.1074/jbc.274.5.2625
Pan T, Kondo S, Le W, Jankovic J. 2008. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 131, 1969-1978.
Park C.et al. (2003) Quercetin protects the hydrogen peroxide-induced apoptosis via inhibition of mitochondrial dysfunction in H9c2 cardiomyoblast cells. Biochem Pharmacol 66(7): 1287-1295.
Park SE, Sapkota K, Choi JH, Kim MK, Kim YH, Kim KM, Kim KJ, Oh HN, Kim SJ, Kim S (2014) Rutin from Dendropanax morbifera Leveille protects human dopaminergic cells against rotenone induced cell injury through inhibiting JNK and p38 MAPK signaling. Neurochem Res. 39(4):707-18.
Parker WD Jr, Boyson SJ, Parks JK. 1989. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol.26(6):719-23.
pasqualiL, Caldarazzo-Ienco Fornai . (2014). MPTP neurotoxicity:actions, mechanisms, and animal modeling of Parkinson's disease. In: Kostrzewa RM (ed) Handbook of neurotoxicity. Springer, Heidelberg, pp237-275.
Peschanski M, Defer G, N'Guyen JP, Ricolfi F, Monfort JC, Remy P, Geny C, Samson Y, Hantraye P, Jeny R. 1994. Bilateral motor improvement and alteration of L-dopa effect in two patients with Parkinson's disease following intrastriatal transplantation of foetal ventral mesencephalon. Brain. 117 ( Pt 3):487-99.
Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS (2001) Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 106(3):589-601.
Powers ET1, Morimoto RI, Dillin A, Kelly JW, Balch WE. 2009.. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Ann. Rev. Biochem 78: 959–91.
Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA. 2007. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis 25(2): 392-400. Parker WD Jr, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. 1189:215-8.
Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, et al. 2011. beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol 186(7): 4443-4454.
Rappold PM et al.2014. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nature Communications. 5:5244 doi: 10.1038/ncomms6244.
Ren Y. et al., 2005. Selectivwe vulnerabity of dopaminergic neurons to microtubule depolymerisation. J. Bio. Chem. 280:434105-12. Reynolds GP, Garrett NJ (1986) Striatal dopamine and homovanillic acid in Huntington's disease. J Neural Transm. 65(2):151-5.
Riederer BM, Pellier V, Antonsson B, Di Paolo G, Stimpson SA, Lütjens R, Catsicas S, Grenningloh G. Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc Natl Acad Sci U S A. 1997 Jan 21;94(2):741-5. doi: 10.1073/pnas.94.2.741. PMID: 9012855; PMCID: PMC19584.
Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. 2010. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia 58(5): 588-598.
Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, et al. 2011. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci U S A 108(16): 6632-6637.
Rossignol R, Malgat M, Mazat JP, Letellier T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J Biol Chem. 1999 Nov 19;274(47):33426-32. doi: 10.1074/jbc.274.47.33426. PMID: 10559224.
Rossignol R, Letellier T, Malgat M, Rocher C, Mazat JP. Tissue variation in the control of oxidative phosphorylation: implication for mitochondrial diseases. Biochem J. 2000 Apr 1;347 Pt 1(Pt 1):45-53. PMID: 10727400; PMCID: PMC1220929.
Rubio-Perez JM, Morillas-Ruiz JM. 2012. A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal 2012: 756357.
Salama M, Helmy B, El-Gamal M, Reda A, Ellaithy A, Tantawy D, et al. 2013. Role of L-thyroxin in counteracting rotenone induced neurotoxicity in rats. Environmental toxicology and pharmacology 35(2): 270-277.
Sandström J, Broyer A, Zoia D, Schilt C, Greggio C1, Fournier M, Do KQ, Monnet-Tschudi F. Potential mechanisms of development-dependent adverse effects of the herbicide paraquat in 3D rat brain cell cultures.Neurotoxicology. 2017 May;60:116-124. doi: 10.1016/j.neuro.2017.04.010. Epub 2017 Apr 30.
Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP. 2006. L-deprenyl protects against rotenone-induced, oxidative stress-mediated dopaminergic neurodegeneration in rats. Neurochem Int.49(1):28-40.
Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al. 2012. S100B is increased in Parkinson's disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain 135(Pt 11): 3336-3347.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, and Marsden CD. 1989. Mitochondrial complex I de. ciency in Parkinson’s disease. Lancet. 1,1269.
Scott R, Gregory R, Hines N, Carroll C, Hyman N, Papanasstasiou V, Leather C, Rowe J, Silburn P, Aziz T. 1998. Neuropsychological, neurological and functional outcome following pallidotomy for Parkinson's disease. A consecutive series of eight simultaneous bilateral and twelve unilateral procedures. Brain. 121 ( Pt 4):659-75.
Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. 2002. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci. 22(16):7006-15.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al. 2003. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 23:10756–64.
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT (2007) Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J Neurochem. 100(6):1469-79.
Shulman JM, DeJager PL, Feany MB. 2011. Parkinson’s disease: Genetics and Pathogenesis. Annu.Rev.Pathol.Mech.Dis. 6:193-2
Shults CW. 2004. Mitochondrial dysfunction and possible treatments in Parkinson’s disease–a review. Mitochondrion 4:641– 648.
Singer TP, Ramsay RR.The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochim Biophys Acta. 1994 Aug 30;1187(2):198-202.
Spencer DD, Robbins RJ, Naftolin F, Marek KL, Vollmer T, Leranth C, Roth RH, Price LH, Gjedde A, Bunney BS. 1992. Unilateral transplantation of human fetal mesencephalic tissue into the caudate nucleus of patients with Parkinson's disease. N Engl J Med. 1992 Nov 26;327(22):1541-8
Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. 2002. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson's disease. Faseb J 16(11): 1474-1476.
Sulzer D, Surmeier DJ. 2013. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Movement Disorders. 28 (6) 715-24.
Surmeier DJ1, Guzman JN, Sanchez-Padilla J, Goldberg JA. 2010. What causes the death of dopaminergic neurons in Parkinson's disease? Prog Brain Res. 2010;183:59-77. doi: 10.1016/S0079-6123(10)83004
Silva MA, Mattern C, Häcker R, Tomaz C, Huston JP, Schwarting RK. 1997. Increased neostriatal dopamine activity after intraperitoneal or intranasal administration of L-DOPA: on the role of benserazide pretreatment. Synapse. 27(4):294-302.
Szibor M, Gainutdinov T, Fernandez-Vizarra E, Dufour E, Gizatullina Z, Debska-Vielhaber G, Heidler J, Wittig I, Viscomi C, Gellerich F, Moore AL. Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim Biophys Acta Bioenerg. 2020 Feb 1;1861(2):148137. doi: 10.1016/j.bbabio.2019.148137. Epub 2019 Dec 9. PMID: 31825809.
Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP Jr, Davis RE, Parker WD Jr (1996) Origin and functional consequences of the complex I defect in Parkinson's disease. Ann Neurol. 40(4):663-71.
Taetzsch T, Block ML. 2013. Pesticides, microglial NOX2, and Parkinson's disease. J Biochem Mol Toxicol 27(2): 137-149.
Tansey MG, Goldberg MS. 2009. Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis.
Tebby C, Gao W, Delp J, Carta G, van der Stel W, Leist M, Jennings P, van de Water B, Bois FY. A quantitative AOP of mitochondrial toxicity based on data from three cell lines. Toxicol In Vitro. 2022 Jun;81:105345. doi: 10.1016/j.tiv.2022.105345. Epub 2022 Mar 10. PMID: 35278637.
Thomas B., Banerjee R.,Starkova NN., Zhang S., Calingasan NY, Yang L., Wille E., Lorenzo B., Ho D., Beal M., Starkov A. 2012. Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson's disease. Antioxidants & redox signaling 16 (9) 855-68
Thundyil J, Lim KL. 2014. DAMPs and Neurodegeneration. Ageing research reviews.
Tieu Kim, Imm Jennifer. 2014. Mitochondrial dynamics as potential therapeutic target for Parkinson’s disease? ACNR 14 (1) 6-8.
Tseng YT, Chang FR, Lo YC2. 2014. The Chinese herbal formula Liuwei dihuang protects dopaminergic neurons against Parkinson's toxin through enhancing antioxidative defense and preventing apoptotic death. Phytomedicine. 21(5):724-33.
Uitti RJ, Ahlskog JE. 1996. Comparative Review of Dopamine Receptor Agonists in Parkinson's Disease. C NS Drugs. 5(5):369-88.
Uitti RJ, Wharen RE Jr, Turk MF, Lucas JA, Finton MJ, Graff-Radford NR, Boylan KB, Goerss SJ, Kall BA, Adler CH, Caviness JN, Atkinson EJ. 1997. Unilateral pallidotomy for Parkinson's disease: comparison of outcome in younger versus elderly patients. Neurology. 49(4):1072-7.
van der Stel W, Carta G, Eakins J, Darici S, Delp J, Forsby A, Bennekou SH, Gardner I, Leist M, Danen EHJ, Walker P, van de Water B, Jennings P. Correction to: Multiparametric assessment of mitochondrial respiratory inhibition in HepG2 and RPTEC/TERT1 cells using a panel of mitochondrial targeting agrochemicals. Arch Toxicol. 2020 Aug;94(8):2731-2732. doi: 10.1007/s00204-020-02849-5. Erratum for: Arch Toxicol. 2020 Aug;94(8):2707-2729. doi: 10.1007/s00204-020-02792-5. PMID: 32720191; PMCID: PMC7645484.
van der Stel, Wanda; Bennekou, Susanne Hougaard; Carta, Giada; Eakins, Julie; Delp, Johannes; Forsby, Anna; Kamp, Hennicke; Gardner, Ian; Zdradil, Barbara; Pastor, Manual (2020) ENV/JM/MONO(2020)22 CASE STUDY ON THE USE OF INTEGRATED APPROACHES TO TESTING AND ASSESSMENT FOR IDENTIFICATION AND CHARACTERISATION OF PARKINSONIAN HAZARD LIABILITY OF DEGUELIN BY AN AOP-BASED TESTING AND READ ACROSS APPROACH Series on Testing and Assessment No. 326
van der Stel W, Yang H, Vrijenhoek NG, Schimming JP, Callegaro G, Carta G, Darici S, Delp J, Forsby A, White A, le Dévédec S, Leist M, Jennings P, Beltman JB, van de Water B, Danen EHJ. Mapping the cellular response to electron transport chain inhibitors reveals selective signaling networks triggered by mitochondrial perturbation. Arch Toxicol. 2022 Jan;96(1):259-285. doi: 10.1007/s00204-021-03160-7. Epub 2021 Oct 13. PMID: 34642769; PMCID: PMC8748354.
Vivekanantham S, Shah S, Dewji R,Dewji A, Khatri C & Ologunde R.2014. Neuroinflammation in Parkinson's disease: role in neurodegeneration and tissue repair. International Journal Of NeuroscienceAccepted. Author Version Posted Online.
Walter BL, Vitek JL. 2004. Surgical treatment for Parkinson's disease. Lancet Neurol. 3(12):719-28.
Widner H, Tetrud J, Rehncrona S, Snow B, Brundin P, Gustavii B, Björklund A, Lindvall O, Langston JW. 1992. Bilateral fetal mesencephalic grafting in two patients with parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N Engl J Med. 26;327(22):1556-63.
walker DG, Lue LF, Serrano G, Adler CH, Caviness JN, Sue LI, Beach T.2016. altered expression patterns of inflammation-associated and trophic molecules in substantia nigra and striatum brain samples from Parkinson's disease, incidental Lewy Body disease and normal control cases. Frontiers in Neuroscience. 9:507.
Wang Q, Chu CH, Oyarzabal E, Jiang L, Chen SH, Wilson B, et al. 2014. Subpicomolar diphenyleneiodonium inhibits microglial NADPH oxidase with high specificity and shows great potential as a therapeutic agent for neurodegenerative diseases. Glia 62(12): 2034-2043.
Wang T, Zhang W, Pei Z, Block M, Wilson B, Reece JM, et al. 2006. Reactive microgliosis participates in MPP+-induced dopaminergic neurodegeneration: role of 67 kDa laminin receptor. Faseb J 20(7): 906-915.
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH (2011) Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 286(18):16504-15.
Wu XF, Block ML, Zhang W, Qin L, Wilson B, Zhang WQ, et al. 2005. The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxidants & redox signaling 7(5-6): 654-661.
Wu RM, Mohanakumar KP, Murphy DL, Chiueh CC. 1994. Antioxidant mechanism and protection of nigral neurons against MPP+ toxicity by deprenyl (selegiline). Ann N Y Acad Sci. 17;738:214-21.
Yadav S, Gupta SP, Srivastava G, Srivastava PK, Singh MP. 2012. Role of secondary mediators in caffeine-mediated neuroprotection in maneb- and paraquat-induced Parkinson's disease phenotype in the mouse. Neurochem Res 37(4): 875-884.
Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A. 2015. Mitochondrial Dysfunction and α-Synuclein Synaptic Pathology in Parkinson's Disease: Who's on First? Parkinsons Dis. 2015:108029.
Zhou F, Wu JY, Sun XL, Yao HH, Ding JH, Hu G. 2007. Iptakalim alleviates rotenone-induced degeneration of dopaminergic neurons through inhibiting microglia-mediated neuroinflammation. Neuropsychopharmacology 32(12): 2570-2580.
Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. 2007.Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 170:75–86.
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
Search strings
Each strategy followed this structure: Stressor AND Parkinson
Parkinson’s Disease
|
PubMed |
("Parkinsonian Disorders"[MeSH Terms:noexp] OR "Parkinson Disease"[MeSH Terms] OR "Lewy Body Disease"[MeSH Terms] OR "lewy bodies/pathology"[MeSH Terms] OR "Synucleinopathies"[MeSH Terms:noexp] OR "Tremor"[MeSH Terms]) OR (Parkinson* [Title/Abstract] OR paralys*-agitans[Title/Abstract] OR Shaking-pals*[Title/Abstract] OR Synuclein*-Patholog*[Title/Abstract] OR synucleinopathol*[Title/Abstract] OR synuclein*-linked-disease*[Title/Abstract] OR synuclein*-linked-neurodegenerat*[Title/Abstract] OR synuclein*-linked-neuro-degenerat*[Title/Abstract] OR synuclein-linked*-oligodendrogliopath*[Title/Abstract] OR TREMOR*[Title/Abstract] OR QUIVER*[TITLE/ABSTRACT] OR ((LEWY*[tiab]) AND (DEMENT*[Title/Abstract] OR DISEASE*[Title/Abstract] OR PATHOL*[TIAB] OR DISORDER*[tiab]))) |
|
EMBASE |
('parkinsonism'/exp OR 'Parkinson disease'/de OR 'synucleinopathy'/de OR 'tremor'/exp OR 'diffuse Lewy body disease'/exp OR 'Lewy body'/exp) OR (Parkinson*:ti,ab,kw OR paralys*-agitans:ti,ab,kw OR Shaking-pals*:ti,ab,kw OR Synuclein*-Patholog*:ti,ab,kw OR synucleinopathol*:ti,ab,kw OR synuclein*-linked-disease*:ti,ab,kw OR synuclein*-linked-neurodegenerat*:ti,ab,kw OR synuclein*-linked-neuro-degenerat*:ti,ab,kw OR synuclein-linked*-oligodendrogliopath*:ti,ab,kw OR TREMOR*:ti,ab,kw OR QUIVER*:ti,ab,kw OR ((LEWY*:ti,ab,kw) AND (DEMENT*:ti,ab,kw OR DISEASE*:ti,ab,kw OR PATHOL*:ti,ab,kw OR DISORDER*:ti,ab,kw))) |
|
Scopus |
( ( ( TITLE-ABS-KEY ( lewy* AND ( dement* OR disease* OR pathol* OR disorder* ) ) ) OR ( TITLE-ABS-KEY ( parkinson* OR paralys*-agitans OR shaking-pals* OR synuclein*-patholog* OR synucleinopathol* OR synuclein*-linked-disease* OR synuclein*-linked-neurodegenerat* OR synuclein*-linked-neuro-degenerat* OR synuclein-linked*-oligodendrogliopath* OR tremor* OR quiver* ) ) ) ) |
|
Web of Science |
TS=((Parkinson* OR paralys*-agitans OR Shaking-pals* OR Synuclein*-Patholog* OR synucleinopathol* OR synuclein*-linked-disease* OR synuclein*-linked-neurodegenerat* OR synuclein*-linked-neuro-degenerat* OR synuclein-linked*-oligodendrogliopath* OR TREMOR* OR QUIVER*) OR (LEWY* AND (DEMENT* OR DISEASE* OR PATHOL* OR DISORDER*)) and Preprint Citation Index (Exclude – Database)) |
Mitochondrial dysfunction
|
PubMed |
((((((oxygen*[Title/Abstract] OR ROS[Title/Abstract] OR oxidative-stress*[Title/Abstract] OR free-radical*[Title/Abstract] OR superoxide*[Title/Abstract] OR hydrogen-peroxide[Title/Abstract] OR peroxyl-radical*[Title/Abstract] OR electron-transport*[Title/Abstract] OR electron-transfer[Title/Abstract] OR ETC[Title/Abstract] OR respirat*[Title/Abstract] OR oxidative-phosphoryl*[Title/Abstract] OR redox[Title/Abstract] OR bioenerget*[Title/Abstract] OR metabol*[Title/Abstract] OR ATP-synth*[Title/Abstract] OR adenosine-triphosphate-synth*[Title/Abstract] OR catabolism*[Title/Abstract] OR anabolism*[Title/Abstract] OR energ*-product*[Title/Abstract] OR Membran*[Title/Abstract] OR transmembran*[Title/Abstract] OR potential*[Title/Abstract] OR permeabil*[Title/Abstract] OR hypox*[Title/Abstract] OR toxic*[Title/Abstract] OR intox*[Title/Abstract] OR poison*[Title/Abstract] OR contaminat*[Title/Abstract] OR hazard*[Title/Abstract] OR adverse-effect*[Title/Abstract] OR deleteri*[Title/Abstract] OR noxious[Title/Abstract] OR harmful[Title/Abstract] OR detrimental[Title/Abstract] OR oxidation[Title/Abstract]) AND (mitochondr*[Title/Abstract] OR mtDNA[Title/Abstract])) OR (((Mitochondr*[Title/Abstract] OR mtDNA[Title/Abstract] OR Megaconial[Title/Abstract] OR Pleoconial[Title/Abstract] OR Carbamyl-Phosphate-Synth*[Title/Abstract] OR Carbamoylphosphate-Synth*[Title/Abstract] OR CarbamylPhosphate-Synth*[Title/Abstract] OR Carbamoyl-phosphate-Synth*[Title/Abstract] OR CPS-I[Title/Abstract] OR CPS-1[Title/Abstract] OR Cytochrome-c-Oxidas*[Title/Abstract] OR Cox[Title/Abstract] OR Cytochrome-Oxidas*[Title/Abstract] OR Complex[Title/Abstract] OR Multiple-Acyl-CoA-Dehydrogenas*[Title/Abstract] OR Electron-Transfer-Flavoprotein*[Title/Abstract] OR ETFA[Title/Abstract] OR ETFB[Title/Abstract] OR ETFDH[Title/Abstract] OR Pyruvate-Carboxylas*[Title/Abstract] OR PDH[Title/Abstract] OR Pyruvate-Dehydrogenas*[Title/Abstract] OR PDHC[Title/Abstract] OR NADH[Title/Abstract] OR succinate-coenzyme-Q[Title/Abstract] OR succinate-CoQ[Title/Abstract] OR succinate-dehydrogen*[Title/Abstract] OR ubiquinol-cytochrom*[Title/Abstract] OR ubiquin*[Title/Abstract] OR respiratory-chain[Title/Abstract] OR Oxidative-Phosphorylat*[Title/Abstract])) AND (Deficien*[Title/Abstract] OR lack*[Title/Abstract] OR decreas*[Title/Abstract] OR reduct*[Title/Abstract] OR diminut*[Title/Abstract] OR deteriorat*[Title/Abstract] OR Diseas*[Title/Abstract] OR disorder*[Title/Abstract] OR dysfunction*[Title/Abstract] OR Defect*[Title/Abstract] OR insufficien*[Title/Abstract] OR inadequac*[Title/Abstract] OR impair*[Title/Abstract] OR loss[Title/Abstract] OR shortag*[Title/Abstract] OR diminish*[Title/Abstract] OR deplet*[Title/Abstract] OR degenerat*[Title/Abstract] OR Myopath*[Title/Abstract] OR Encephalomyopath*[Title/Abstract] OR syndrom*[Title/Abstract] OR condition*[Title/Abstract] OR abnormal*[Title/Abstract] OR patholog*[Title/Abstract] OR cytopath*[Title/Abstract] OR malfunction*[Title/Abstract] OR anomal*[Title/Abstract] OR damag*[Title/Abstract] OR shrink*[Title/Abstract] OR atroph*[Title/Abstract] OR injur*[Title/Abstract] OR compromis*[Title/Abstract] OR disturb*[Title/Abstract] OR fail*[Title/Abstract] OR breakdown[Title/Abstract] OR declin*[Title/Abstract] OR weaken*[Title/Abstract] OR fragilit*[Title/Abstract] OR instabil*[Title/Abstract]))) OR (Subacute-Necrotizing-Encephalomyel*[Title/Abstract] OR Subacute-Necrotizing-Encephalopath*[Title/Abstract] OR CPEO[Title/Abstract] OR Glutaric-Acidur*[Title/Abstract] OR MADD[Title/Abstract] OR MADDs[Title/Abstract] OR methylmalonic-acidur*[Title/Abstract] OR Ethylmalonic-Adipic-Acidur*[Title/Abstract] OR Ethylmalonic-Adipicacidur*[Title/Abstract] OR Glutaric-Acidem*[Title/Abstract] OR Abnormal-Pyruvate-Metabolism[Title/Abstract] OR Lactic-Acidosis[Title/Abstract] OR mtODE[Title/Abstract] OR oxidative-damage-specific-endonucl*[Title/Abstract] OR MTP[Title/Abstract] OR MOMP[Title/Abstract] OR mitochondriopath*[Title/Abstract] OR MNGIE[Title/Abstract])) OR (Alper*-disease*[Title/Abstract] OR Alper*-syndrom*[Title/Abstract] OR Alpers-Huttenlocher-diseas*[Title/Abstract] OR Alpers-Huttenlocher syndrom*[Title/Abstract])) OR (progressive[Title/Abstract] AND poliodystrophy*[Title/Abstract])) OR (((("mitochondrial oxidative damage endonuclease" [Supplementary Concept]) OR "Oxygen Consumption"[Mesh]) OR "Mitochondrial Diseases"[Mesh]) OR "Membrane Potential, Mitochondrial"[Mesh]) (Oxygen-Consumpt*[Title/Abstract] OR Anaerobic-Threshold*[Title/Abstract] OR Metabolic-Equivalent*[Title/Abstract]) |
Stressors
|
Deguelin |
|
|
PubMed |
("deguelin" [Supplementary Concept] or deguelin*[tiab]) |
|
EMBASE |
('deguelin'/exp OR deguelin*:ti,ab,kw,tn) |
|
Scopus |
( TITLE-ABS-KEY ( deguelin ) ) |
|
Web of Science |
(TS=(deguelin) |
|
Fenpyroximate |
|
|
PubMed |
("fenpyroximate" [Supplementary Concept] or fenpyroximat*[tiab]) |
|
EMBASE |
('fenpyroximate'/exp OR fenpyroximat*:ti,ab,kw,tn) |
|
Scopus |
( TITLE-ABS-KEY ( fenpyroximat* ) ) |
|
Web of Science |
TS=(fenpyroximat*) |
|
Pyrimidifen |
|
|
PubMed |
"pyrimidifen" [Supplementary Concept] OR Pyrimidifen[tiab] |
|
EMBASE |
Pyrimidifen:ti,ab,kw,tn |
|
Scopus |
TITLE-ABS-KEY ( piyrimidifen* ) ) |
|
Web of Science |
TS=(pyrimidifen*) |
|
Tebufenpyrad |
|
|
PubMed |
("4-chloro-N-((4-(1,1-dimethylethyl)phenyl)methyl)-3-ethyl-1-methyl-1H-pyrazole-5-carboxamide" [Supplementary Concept] OR Tebufenpyrad[TIAB] OR MK239[TIAB]) |
|
EMBASE |
('tebufenpyrad'/exp OR (tebufenpyrad:ti,ab,kw,tn OR mk239:ti,ab,kw,tn)) |
|
Scopus |
( TITLE-ABS-KEY ( Tebufenpyrad* OR mk239 ) ) |
|
Web of Science |
(TS=(Tebufenpyrad* OR mk239) |