
This Key Event Relationship is licensed under the Creative Commons BY-SA license. This license allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use. If you remix, adapt, or build upon the material, you must license the modified material under identical terms.
Relationship: 934
Title
Inhibition, NADH-ubiquinone oxidoreductase (complex I) leads to Increase, Mitochondrial dysfunction
Upstream event
Downstream event
Key Event Relationship Overview
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding | Point of Contact | Author Status | OECD Status |
|---|---|---|---|---|---|---|
| Inhibition of the mitochondrial complex I of nigro-striatal neurons leads to parkinsonian motor deficits | adjacent | High | Moderate | Andrea Terron (send email) | Open for citation & comment | WPHA/WNT Endorsed |
Taxonomic Applicability
Sex Applicability
| Sex | Evidence |
|---|---|
| Male | High |
| Female | High |
Life Stage Applicability
| Term | Evidence |
|---|---|
| All life stages | High |
Key Event Relationship Description
Inhibited CI is unable to pass off its electron to ubiquinone and it cannot translocate protons across the mitochondrial inner membrane. This creates a back-up of NADH within the mitochondrial matrix (Brown and Borutaite, 2004). This leads to an arrest of the citric acid cycle and a failure to build a proton gradient (mitochondrial membrane potential, Δψm) across the inner membrane. This results in impaired ATP production. In addition, the direct transfer of electrons from CI to oxygen is increased. This leads to oxidative stress as ROS (e.g. superoxide, hydrogen peroxide) are produced, which can damage DNA, proteins, lipids and other cell components and function (Sanders et al., 2014).
Evidence Collection Strategy
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
The implementation of AOP3 is based on a negotiated procedure with EFSA (reference NP/EFSA/PREV/2024/02). This procedure is intended to update AOP3 by adding more evidence to the AOP Wiki, considering the contribution of mitochondrial complex I for which a strong biological plausibility that its inhibition leads to degeneration of dopaminergic neurons exist. The starting conceptual model for this project is based on the key scientific sources, including EFSA (2017), Delp et al. (2019 and 2021), Van der Stel et al. (2020 and 2022), ENV/JM/MONO(2020)22, Alimohammadi et al. (2023), Tebby et al (2022). These publications provided the initial basis for this project and contributed to the Empirical Evidence.
Additional literature was identified through a structured, non systematic search using a stressor-based search strategy as described in the “AOP development strategy” section.
- Not endorsed
Evidence Supporting this KER
The weight of evidence supporting the relationship between inhibition of CI and mitochondrial dysfunction is strong. The mechanisms behind this KER are partially understood and well documented based on in vitro as well as in vivo experiments (e.g., Sanders et al., 2014), complemented by data from human post-mortem PD brain evaluations (Parker et al., 1989; Greenamyre et al., 2001; Sherer et al., 2003; Schapira et al., 1989).
Biological Plausibility
The biological plausibility that inhibition of CI activity triggers mitochondrial dysfunction is strong. It is well understood, how the inhibition of CI can lead to mitochondrial dysfunction as measured by: a) decreased oxygen consumption, b) decrease or loss of ATP production, c) decrease of Δψm, d) the loss of mitochondrial protein import and protein biosynthesis, e) reduced activities of enzymes of the mitochondrial respiratory chain and the Krebs cycle, f) elevated levels of ROS, g) the loss of mitochondrial motility, causing a failure of mitochondria to re-localize to sites of increased energy demands (such as synapses), h) destruction of the mitochondrial network, i) increased mitochondrial uptake of Ca2+ causing mitochondrial Ca2+ overload (Graier et al., 2007) and opening of mitochondrial PTP, (j) rupture of the mitochondrial inner and outer membranes, leading to release of mitochondrial pro-death factors, including cytochrome c, AIF and endonuclease G (Braun, 2012; Martin, 2011; Correia et al., 2012; Cozzolino et al., 2013). These pathological mechanisms are extremely well studied.
Empirical Evidence
Many studies show that the pathophysiological consequences of a partial or total CI inhibition are linked to mitochondrial dysfunction. In many of these experiments the cellular damage caused by mitochondrial dysfunction is reduced (or entirely prevented) by treatment with antioxidants. Different degrees of CI inhibition by rotenone have been studied in the human osteosarcoma-derived cell line (143B). A quantitative correlation between increasing inhibition of CI and mitochondrial dysfunction (as shown by inhibition of mitochondrial respiration, reduced ATP production, increased ROS release and lipid peroxidation, as well as decreased Δψm) was established (Fig. 1 and Table 1 based on Barrientos and Moraes, 1999). Based on the existing literature it is suggested that rotenone exerts toxicity via oxidative stress, rather than via decrease of ATP synthesis (bioenergetics effects).
A few examples illustrating mitochondrial damage and oxidative stress in animal model of PD and human cells induced by:
Rotenone
- Rotenone administered subcutaneously for 5 weeks (2.5 mg/kg/d) caused a selective increase (by ~2 folds) in oxidative damage in the striatum, as compared to the hippocampus and cortex, accompanied by massive degeneration of DA neurons (~80% decrease) in the substantia nigra. Rotenone reduced intracellular ATP levels in the striatum (by >40%), increases malondialdehyde (MDA, indicative of lipid peroxidation, by ~60%), reduced GSH levels (by ~20%), thioredoxin (by ~70%), and manganese superoxide dismutase (SOD, by ~15%) (all parameters significantly changed in the striatum). Antioxidant polydatin (Piceid) treatment significantly prevented the rotenone-induced changes by restoring the above parameters to control levels, confirming that rotenone- induced mitochondrial dysfunction resulted in oxidative stress (Chen et al., 2015).
- Rotenone was administered 2.5 mg/kg body weight to male Wistar rats for 4 weeks in the presence or absence of ferulic acid (FA, at the dose of 50 mg/kg) that has antioxidant and anti-inflammatory properties. Rotenone administration caused DA neuronal cell death (~50%), significant reduction in endogenous antioxidants, such as superoxide dismutase (~75%), catalase (~40%), and glutathione (~50%), and induced lipid peroxidation evidenced by increased MDA formation (~2 folds). Treatment with FA rescued DA neurons in substantia nigra pars compacta area and nerve terminals in the striatum, as well as restored antioxidant enzymes, prevented depletion of glutathione, and inhibited lipid peroxidation induced by rotenone (Ojha et al., 2015).
- Many studies have shown that mitochondrial aldehyde dehydrogenase 2 (ALDH2) functions as a cellular protector against oxidative stress by detoxification of cytotoxic aldehydes. Dopamine is metabolized by monoamine oxidase to yield 3,4-dihydroxyphenylacetaldehyde (DOPAL) then converts to a less toxic acid product by ALDH. The highly toxic and reactive DOPAL has been hypothesized to contribute to the selective neurodegeneration of DA neurons. In this study, rotenone (100 nM, 24 hr) in both SH-SY5Y cells and primary cultured substantia nigra (SN) DA neurons, was shown to reduce DA cell viability (~40%), reduce Δψm (~40%, as shown by TMRM), induce mitochondrial ROS production (~30%, as shown by increase of MitoSox Red), and increased cytosolic protein levels of proteins related to the mitochondrial apoptotic pathway (i.e. Bax, cytochrome c, active caspase-9 and active caspase-3) (~ 2 folds for all proteins).
The neuroprotective mechanism of ALDH2 was observed as overexpression of wild-type ALDH2 gene (but not the enzymatically deficient mutant ALDH2*2 (E504K)) reduced rotenone-induced DA neuronal cell death, prevented rotenone-induced reduction in TMRM signal (95.7±1.6% v.s. 67±3.5%), and prevented rotenone-induced increase in MitoSox Red intensity (103.1±1% v.s. 133.4±0.8%). Additionally, pre-treatment of cells with Alda-1 (activator of ALDH2) (1–10 μM, for 24 hr) prevented rotenone-induced loss of Δψm and ROS production in a dose-dependent manner. These results were confirmed by in vivo studies. Rotenone (50 mg/kg/day, oral administration for 14 days) or MPTP (40 mg/kg/day, i.p. for 14 days) both administered to C57BL/6 mice caused significant SN TH+ DA neuronal cell apoptosis (~50%). Alda-1 attenuated rotenone-induced apoptosis by decreasing ROS accumulation, reversing Δψm depolarization, and inhibiting the activation of proteins related to mitochondrial apoptotic pathway. The present study demonstrates that rotenone or MPP+ induces DA neurotoxicity through oxidative stress. Moreover, Alda-1 is effective in ameliorating mitochondrial dysfunction by inhibiting rotenone or MPP+ induced mitochondria-mediated oxidative stress that leads to apoptosis (Chiu et al., 2015).
- Rotenone-induced mitochondrial dysfunction was observed in human neuroblastoma cells exposed to 5 nM rotenone for 1-4 weeks. After 3-4 weeks of treatment, rotenone-treated cells showed evidence of oxidative stress, including loss of GSH (by 5%) and increased oxidative DNA (qualitative, measured by using antibodies to 8-oxo-dG) and protein damage (223 ± 29% of control, as shown by the large increase in protein carbonyls in the insoluble fraction) (Sherer et al. 2002). This chronic rotenone treatment markedly sensitized cells to further oxidative challenge since in response to H2O2 cytochrome c release from mitochondria and caspase-3 activation occurred earlier and to a greater extent in rotenone-treated cells vs Ctr (1.44 ± 0.02% vs 0.38 ± 0.07% apoptosis/hr). This study indicates that chronic, low-level CI inhibition by rotenone induces progressive oxidative damage, and caspase-dependent neuronal cell death (Sherer et al., 2002).
- By using anti-oxidant, kaempferol (6 μM, 1 hr prior addition of rotenone) and rotenone (50 nM, max up to 24 hr) on SH-SY5Y cells, kaempferol was found to counteract rotenone-induced ROS production (especially superoxide: with kaempferol, ethidium fluorescence decreased below the control (Ctr) levels), rotenone-induced mitochondrial oxidative dysfunction (protein carbonyls values: 2.5 in Ctr, 6.2 with rotenone, 2.7 with kaempferol + rotenone), rotenone-induced oxygen respiration (values of nmol of atomic oxygen/minute/mg protein: 5.89 Ctr, 0.45 with rotenone, 2.47 with kaempferol + rotenone), rotenone-induced Δѱm decrease (~70% cells of with rotenone only vs ~30% with kaempferol + rotenone) (Filomeni et al., 2012).
- To model the systemic mitochondrial impairment, rats were exposed to rotenone. A single rotenone dose (10 nM, for 24 hr) induced mtDNA damage in midbrain neurons (>0.4 lesions/10kb vs 0 lesions/10kb in vehicle), but not in cortical neurons; similar results were obtained in vitro in cultured neurons. Importantly, these results indicate that mtDNA damage is detectable prior to any signs of neuronal degeneration and is produced selectively in midbrain neurons. The selective vulnerability of midbrain neurons to mtDNA damage was not due to differential effects of rotenone on CI since rotenone suppressed respiration equally (~60%) in midbrain and cortical neurons compared to vehicle. However, in response to CI inhibition, midbrain neurons produced more mitochondrial H2O2 (5 min of rotenone increased MitoPY1 fluorescence of ~10% in midbrain mitochondria vs vehicle, and progressively for the duration of measurement), than cortical neurons. The selective mtDNA damage in midbrain could serve as a molecular marker of vulnerable nigral neurons in PD. Oxidative damage to cell macromolecules in human PD and the rotenone model have been recently reviewed (Sanders et al., 2014).
- Adult male Sprague–Dawley rats were intranigrally infused with rotenone (6 μg in 1 μl) alone or in the presence of L-deprenyl (0.1, 1, 5 and 10 mg/kg; i.p.) at 12 h intervals for 4 days. Rotenone alone (100 μM, 30 min) increased the levels of hydroxyl radials in the mitochondrial P2 fraction 2,3-DHBA (122.90 ± 5.4 pmol/mg protein) and 2,5-DHBA (146.21 ± 6.3 pmol/mg protein). L-deprenyl (100 nM–1 mM) dose-dependently attenuated rotenone-induced ·OH generation in the mitochondrial P2 fraction. L-deprenyl-induced attenuation in the rotenone-mediated 2,3-DHBA generation was from 17 ± 1.1% to 67 ± 4.3%, respectively, for 100 nM–1 mM of the MAO-B inhibitor. Also, rotenone caused about 51 ± 3.3% reduction in GSH levels in the cell body region, SN and 34 ± 1.1% decrease in the nerve terminal region, NCP (nucleus caudatus putamen). L-deprenyl alone did not cause any significant difference in the GSH content in either region. L-deprenyl treatment dose-dependently attenuated the rotenone-induced GSH depletion in SN from 51 ± 3.1% to 44 ± 2.1%, 32 ± 1.7% and 9 ± 1.0%, respectively, for doses of 1, 5 and 10 mg/kg. Additionally, SOD activity was assayed in rotenone-lesioned animals, which were treated with l-deprenyl at different doses (1–10 mg/kg). SN exhibited 2- and 3-fold activity of Cu/Zn-SOD (i.e. cytosolic SOD fraction) and Mn-SOD (i.e. particulate SOD fraction), respectively, compared to the nerve terminal region, NCP. L-deprenyl (5 and 10 mg/kg) in rotenone-lesioned animals caused a significant increase in the cytosolic Cu/Zn SOD activity in SN of both the sides. Intranigral infusion of rotenone alone caused a significant increase in the enzyme activity in SN of the side of infusion as compared to the non-infused side (~20%). L-deprenyl (5 and 10 mg/kg) further increased catalase activity in both ipsilateral SN and striatum, as compared to the contralateral side of infusion. Finally, rotenone caused a 74% reduction in the striatal TH staining intensity, which was partially recovered by L-deprenyl. These results showed that oxidative stress is one of the major causative factors underlying DA neurodegeneration induced by rotenone and they support the view that L-deprenyl is a potent free radical scavenger and an antioxidant (Saravanan et al., 2006). Similar results were obtained after exposure to MPP+ (Wu et al., 1994).
- Antioxidant (Piperaceae; PLL) with some anti-inflammatory activities demonstrated in preclinical studies protective effects in PD animal models. Rats treated with rotenone and PLL-derived alkaloids showed decreased ROS, stabilized Δψm, and the opening of the mitochondrial PTP - which is triggered by ROS production - was inhibited. In addition, rotenone-induced apoptosis was abrogated in the presence of these alkaloids (Wang H. et al., 2015).
- In SK-N-MC human neuroblastoma cells, rotenone (10 nM - 1µM, 48 hr) caused dose-dependent ATP depletion (~35% reduction by 100 nM rotenone vs Ctr), oxidative damage (100% increase of carbonyls levels upon 100 nM rotenone), and death (100 nM rotenone after 48 hr caused 1.1 AU (arbitrary units) increase of cell death vs untreated – 0.00 AU -). α-Tocopherol pre-treatment (62.5 or 125 μM 24 hr before rotenone (10 nm)) attenuated rotenone toxicity (Sherer et al., 2003).
MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) or MPP+ (1-methyl-4-phenyl-pyridinium ion)
- MPTP converted into MPP+ inhibits mitochondrial CI activity, resulting in excessive intracellular ROS production followed by further mitochondrial dysfunction leading to mitochondrial-dependent apoptosis. Lutein, a carotenoid of xanthophyll family (antioxidant) reversed MPTP-induced mitochondrial dysfunction, oxidative stress, apoptotic cell death and motor abnormalities. These results revealed that antioxidant protected DA neurons and diminished mitochondrial dysfunction and apoptotic death (Nataraj et al., 2015).
- Antioxidant (salidroside; Sal) pre-treatment protected DA neurons against MPTP/MPP+ induced toxicity in a dose-dependent manner by: (1) reducing the production of ROS, (2) regulating the ratio of Bcl-2/Bax, (3) decreasing cytochrome-c and Smac release, and inhibiting caspase-3, caspase-6, and caspase-9 activation, which are known to trigger apoptosis following mitochondrial dysfunction. Sal acted as an effective neuroprotective agent through modulation of the ROS-induced mitochondrial dysfunction in vitro and in vivo (Wang S. et al., 2015).
- In an in vitro study, MPP+ (1 mM, 24 hr) was found to elicit production of ROS (by 2 fold vs Ctr) and reduce by 50% SOD (by about 50%) and catalase (by about 65%) activity in SH-SY5Y human neuroblastoma cells. Pre-treatment with the antioxidant astaxanthin (AST; 50 μM, 24 hr) inhibited MPP+- induced production of ROS and attenuated both SOD and catalase activity decrease. Furthermore, MPP+ (1 mM, 48 hr) increased caspase-3 activity to 243% of the Ctr and also increased cleaved caspase-3 in the cells (qualitative). Addition of 50 μM AST attenuated MPP+-induced caspase-3 activation (57% suppression). MPP+ induced also a 70% reduction of Δψm and cytochrome c release (qualitative), while AST prevented both these effects. The protective effects of AST on MPP+ induced mitochondrial dysfunction was due to its anti-oxidative properties and anti-apoptotic activity via induction of expression of SOD and catalase (as shown above) and regulating the expression of Bcl-2 and Bax (Bax/Bcl-2 ratio increased to 1.6-fold vs Ctr upon treatment with MPP+, while AST prevented the MPP+-induced increase of the Bax/Bcl-2 ratio). These results were confirmed by in vivo studies (Lee et al., 2011).
- DA neurons in primary mesencephalic cultures treated with MPP+ (100 μМ, for 48 hr) underwent reduction of cell viability (~55% MTT reduction), LDH release (~90%), about 60% reduction of TH+ cells, disruption of Δψm (~45% decline) and ROS production (~60% increase), upregulation of Nox2 (~45%) and Nox4 (~60%), while promoting a decrease of both SOD (~45%) and GSH activity (~85%). Additionally, MPP induced apoptosis via mitochondrial dysfunction, as shown by induction of cytochrome c (~55%), cleaved-caspase-3 (~75%), upregulation of Bax expression (~55%), and downregulation of Bcl2 (~60%). Liuwei dihuang (LWDH), a widely used traditional Chinese medicine (TCM), has antioxidant characteristics. LWDH-WH, derivative of LWDH (0.01-10 μg/ml, added 1 hr prior to MPP+ addition) reduced oxidative damage via increasing antioxidant defence (SOD, GSH), decreasing ROS production, and down-regulating NADPH oxidases (Nox2 and Nox4). LWDH-WH also inhibited neuronal apoptosis by increasing anti-apoptotic protein Bcl-2 expression, and down-regulating apoptotic signalling (Bax, cytochrome c, cleaved-caspase-3) in MPP+-treated neurons. All these protective effects were induced in a dose-dependent manner (Tseng et al., 2014).
- PC12 cells treated with MPP+ (500 μM, for 24 hr) underwent reduction of viability (~55% MTT reduction), oxidative stress (~160% increase in ROS production) and downregulation of heme oxygenase-1 expression (~ 2 folds). Pre-treatment with edaravone, a novel free radical scavenger, (25, 50, 75, 100 μM, for 1 h prior MPP+ treatment) protected PC12 cells against MPP+-cytoxicity via inhibiting oxidative stress and up-regulating heme oxygenase-1 expression in a dose-dependent manner (Cheng et al., 2014).
- The protective effects of antioxidant, apigenin (AP), naturally occurring plant flavonoids were observed on the MPP+ induced cytotoxicity in cultured rat adrenal pheochromocytoma cells (PC12 cells). The PC12 cells were pre-treated with various concentrations of the test compound for 4 h, followed by the challenge with 1,000 µM MPP+ for 48 h. Pre-treatment with AP (3 - 6 - 12 µM) before MPP+ significantly reduced the level of intracellular ROS and elevated Δψm in the MPP+ treated PC12 cells. In addition, AP markedly suppressed the increased rate of apoptosis and the reduced Bcl 2/Bax ratio induced by MPP+ in the PC12 cells. The findings demonstrated that AP exerts neuroprotective effects against MPP+ induced neurotoxicity in PC12 cells, at least in part, through the inhibition of oxidative damage and the suppression of apoptosis through the mitochondrial pathway (Liu et al., 2015).
- Brain mitochondria isolated from ventral midbrain of mitochondrial matrix protein cyclophilin D (CYPD) knockout mice were significantly less sensitive to acute MPP+ (20 µM) -induced effects. CYPD ablation attenuated in vitro Ca2+-induced mitochondrial dysfunction and ROS generation upon Ca2+ loading, both in the absence and in the presence of MPP+, compared to wild-type mice. CYPD ablation conferred a protection to mitochondrial functions upon in vivo treatment with MPTP.
Ventral midbrain mitochondria (that constitutes < 5% of SNpc DA neurons) isolated from brains of wild type (wt) mice acutely treated with MPTP (single MPTP 20 mg/kg injection, analysis done after 4 hr), as compared with saline-treated mice, showed a reduction of CI (by 53%), a reduced rate of phosphorylating respiration (by 38%), a reduced respiratory control index (by 37%), and a decreased ADP/O ratio (by 18%). Ventral midbrain mitochondria isolated from brains of CYPD knockout mice acutely treated with MPTP, as compared with MPTP-treated wt mice, exhibited higher activity of CI (~80%, vs 53% wt), higher rate of phosphorylating respiration (~82%, vs 62% wt), a better respiratory control index (~79%, vs 63% wt), and a higher ADP/O ratio (~90% vs 82% wt) (Thomas et al., 2012). CYP plays as a regulatory component of a calcium-dependent permeability transition pores (PTP), and the data suggest that PTP is involved in MPP+-induced mitochondrial damage. Under oxidative stress, the prolonged opening of the PTP results in calcium overload and with time mitochondrial dysfunction as they get de-energized, depolarized, triggering apoptotic or necrotic cell death (Bernardi, 1999).
There are many other studies showing that MPP+ induces NADH-dependent SOD formation and enhances NADH-dependent lipid peroxidation in submitochondrial particles, confirming that oxidative stress is induced by MPP+ (e.g. Takeshige, 1994; Ramsay and Singer, 1992).
Based on the human post mortem studies of PD brains it is well established that oxidative stress and mitochondrial dysfunction accompany the pathophysiology of PD (e.g. Dias et al., 2013; Zhu and Chu, 2010; Hartman et al., 2004; Fujita et al., 2014).
Examples of human data confirming the presence oxidative stress and mitochondrial dysfunction in PD post mortem brains:
- A significant decrease in CI activity has been identified in a large study of post-mortem PD brains, specifically in substantia nigra compared with age matched controls. In idiopathic PD all 10 patients studied had significant reductions of CI activity (Parker et al., 1989). It is hypothesize that the CI dysfunction may have an etiological role in the pathogenesis of PD (Greenamyre et al., 2001; Sherer et al., 2003, Schapira et al., 1989).
- The structure and function of mitochondrial respiratory-chain enzyme proteins were studied post-mortem in the substantia nigra of nine patients with PD and nine matched controls. Total protein and mitochondrial mass were similar in the two groups. CI and NADH cytochrome c reductase activities were significantly reduced, whereas succinate cytochrome c reductase activity was normal. These results indicated a specific defect of CI activity in the substantia nigra of patients with PD (Schapira et al., 1990).
- Post mortem human studies show that CI deficiency in PD is anatomically specific for the substantia nigra, and they are not present in another neurodegenerative disorder involving the substantia nigra. These results suggest that CI deficiency may be the underlying cause of DA cell death in PD (Schapira et al., 1990; Schapira, 1994).
- The mitochondrial respiratory chain function was studied in various brain regions as well as in skeletal muscle and in blood platelets from patients with idiopathic PD and from matched controls. The evidence suggests that the CI deficiency in PD is limited to the brain and that this defect is specific for the substantia nigra (Mann et al., 1992).
- Immunoblotting studies on mitochondria prepared from the striata of patients who died of PD were performed using specific antisera against Complexes I, III and IV. In 4 out of 5 patients with PD, the 30-, 25- and 24-kDa subunits of CI were moderately to markedly decreased. No clear difference was noted in immunoblotting studies on subunits of Complexes III and IV between the control and PD. The authors claim that deficiencies in CI subunits seem to be one of the most important clues to elucidate pathogenesis of PD (Mizuno et al., 1989).
- Redox markers have been found unchanged in PD patient-derived vs Ctr-derived fibroblasts at baseline. Basal mitochondrial respiration and glycolytic capacity resulted similar at baseline between PD and Ctr fibroblasts, while rotenone-sensitive respiration (analysed by using 0.5 μM rotenone) resulted lower in PD fibroblasts vs Ctr (174.74 ± 48.71 vs 264.68 ± 114.84) (Ambrosi et al., 2014).
- Augmented oxidative metabolism has been detected in PD brains by magnetic resonance studies, in conjunction with energy unbalance. Decreased glucose consumption (22% mean reduction), likely reflecting a decrease in neuronal activity, has been reported in the nigrostriatal system of PD patients (Piert et al., 1996). These symptoms were hypothesized to be indicative of mitochondrial dysfunction as early markers, present in the brain of patients with PD even in the absence of overt clinical manifestations (Rango et al., 2006). In particular, by using high temporal and spatial resolution 31P magnetic resonance spectroscopy (31P MRS) technique authors studied mitochondrial function by observing high-energy phosphates (HEPs) and intracellular pH in the visual cortex of 20 PD patients and 20 normal subjects at rest, during, and after visual activation. In normal subjects, HEPs remained unchanged during activation, but rose significantly (by 16%) during recovery, and pH increased during visual activation with a slow return to rest values. In PD patients, HEPs were within the normal range at rest and did not change during activation, but fell significantly (by 36%) in the recovery period; pH did not reveal a homogeneous pattern with a wide spread of values. Energy unbalance under increased oxidative metabolism requirements, that is, the post-activation phase, discloses a mitochondrial dysfunction that is present in the brain of patients with PD even in the absence of overt clinical manifestations,(Rango et al., 2006).
There are many other studies providing evidence that oxidative stress and mitochondrial dysfunction play an important role in PD pathophysiology (see indirect KER Mitochondrial dysfunction induced DA neuronal cell death of nigrastriatal pathway).
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
Data were retrieved from assays measuring endpoints relevant for AOP3 in relevant cell types.
KE 887 Complex I inhibition
Assays
For KE 887, only two assays were employed in the abovementioned publications, both using the Seahorse technology.
• The “LUHMES MitoComplexes” assay (Delp 2019, Delp 2021) used proliferating LUHMES cells.
• The “HepG2 MitoComplexes” assay (van der Stel 2020) uses HepG2 cells.
In both assays, the cells are permeabilized and treated acutely with the test chemical. Oxygen consumption rate (OCR) is then measured under different conditions of active or blocked mitochondrial complexes. Of note, the two assays are setup slightly differently, so that in the LUHMES assay activity on c1 – c4 can be distinguished separately, while in the HepG2 assay, activity on c2 and c3 is measured concurrently.
Cell Models
Despite HepG2 being a hepatic cell line, the assay was included in the evaluation. It was assumed that because the exposure is acute, in permeabilized cells, that the test chemical would have immediate access to the mitochondria. Other mechanisms such as transport into the cells or an indirect effect via other signaling pathways were considered negligible under these assay conditions.
Effect thresholds
The “LUHMES MitoComplexes” assay has an established prediction model (Delp 2019); a chemical is considered active, if OCR is reduced by more than 25% compared to the solvent control.
For the “HepG2 MitoComplexes”, no prediction model has been established. But van der Stel 2020 reported EC50 values, which was used also in the current evaluation.
Table 1. KE 887: criteria for data analysis
|
LUHMES |
HepG2 |
Conclusion |
|
Active* on complex cI |
Active* on complex cI |
strong evidence for KE 887 |
|
Active* on complex cI |
not measured |
evidence for KE 887, with uncertainty. |
|
Active* on complex cI |
inactive or active* on complex other than cI |
evidence for KE 887, with uncertainty. Unless a rationale why there is cell-type specific effects exist |
|
Inactive* |
Active* on complex cI |
Additional evidence needed (update according to OC/EFSA/PREV/2023/01#) |
|
Inactive* |
not measured |
Excluded from the analysis |
|
not measured |
Active* on complex cI |
low evidence for KE 887. Additional evidence needed (update according to OC/EFSA/PREV/2023/01#) |
|
not measured |
Inactive* |
Excluded from the analysis |
*based on Effect threshold, # Environmental Neurotoxicants – Advancing Understanding on the Impact of Chemical Exposure on Brain Health and Disease: Parkinsonian NeurodegenerationRapid Assessment using NAMs
KE177 Mitochondrial dysfunction
Assays
The following assay endpoints were included:
1. Oxygen consumption rate (in intact cells). This was considered the most reliable method to measure a direct effect on mitochondrial respiration.
2. Image-based measurement of mitochondrial membrane potential (MMP). These methods use dyes such as TMRE or rhodamine123.
3. Measurement of ATP levels. This was considered a more indirect measurement of mitochondrial function, as many cell types can generate ATP via glycolysis in glucose medium, which is independent of mitochondrial function.
The following assay endpoints were deemed not suitable for KE177:
• Resazurin: Indirect measure for KE177, the bioreduction of the dye depends on different sources among which mitochondria
• Lactate dehydrogenase release: Indirect measure for KE177, linked to cell viability
• Extracellular acidification rate: acidification/elevation of glycolysis can also be the consequence of an elevated energy demand that can no longer be met by mitochondria. Under these conditions, an elevation of acidification does not correlate with dysfunction of mitochondria
Cell Models
Only neuronal cell models were considered, which included the LUHMES and the SH-SY5Y cells, as relevant for AOP3.
Stressors identification
Chemicals data were extracted from Delp et al. (2019, 2021), Bennekou- ENV/JM/MONO(2020)23, van der Stel- ENVJMMONO(2020)22, van Der Stel et al. (2020), Tebby et al. (2022). Data were carefully evaluated by subject-experts to determine whether the chemical affects KE1542, KE177 and KE890. 5 cI and 4 cIII inhibitors with evidence of interference with KE 887, KE177, according to the criteria described in the previous section, and KE890 were included as stressors in the assessment and subsequently described. An overview of these data across AOPs and KEs, summarising the percentage effect on each KE, is presented in the “Evidence assessment” and "Quantitative understanding" sections of AOP 3 (cI inhibitors).
cI inhibitors
Deguelin
KE 887: Four studies conducted in two cell lines (HepG2, LUHMES) measured inhibition of cI immediately after treatment of cells with deguelin. Two studies in proliferating LUHMES cells were conducted only at a single, high concentration ( 50 µM). Effective concentrations were in the range of 60 – 100 nM for LUHMES and HepG2 cells (Tebby 2022). Of note, these experiments were conducted in permeabilized cells.
KE 177: Multiple studies measured the OCR in differentiating LUHMES immediately after treatment with deguelin. The effective concentrations range from 0,016 µM to 10 µM (Delp 2021, Alimohammadi 2023, Tebby 2022). When cells were treated for 24 h, ATP content was affected in 8-10 µM when cells were cultured in glucose containing medium (ENVJMMONO(2020)22, Delp 2021). This value dropped to 0,0045 µM (2000-fold) when cells were cultured in galactose containing medium (ENVJMMONO(2020)22,).
In differentiated SH-SY5Y cells, 24 h exposure to deguelin lead to a decrease in mitochondrial membrane potential as measured via rhodamine 123 with an EC25 of 0,31 µM, but ATP content was not affected up to 10 µM, which was the highest tested concentration. A prolonged exposure to deguelin (120 h) however, affected the ATP content with an EC50 of 1 µM.
|
KE |
cell type |
assay |
exposure |
effect level |
effect conc |
reference |
notes |
|
KE 887 |
LUHMES (proliferating) |
Mito Complexes, inhibition of c1 |
immediate |
25% |
50 µM (single conc) |
Delp_2019, Suppl. Item 5 |
1 |
|
KE 887 |
LUHMES (proliferating) |
Mito Complexes, inhibition of c1 |
immediate |
25% |
50 µM (single conc) |
Delp_2021, Figure S8 |
1 |
|
KE 887 |
LUHMES (proliferating) |
Mito Complexes, inhibition of c1 |
immediate |
EC25 EC50 |
~ 0,06 µM 0,086 µM |
Tebby 2022, Fig 4 + Table S2 ENVJMMONO(2020)22 van der Stel, Figure 4+5 |
1, 2, 3,4 |
|
KE 887 |
HepG2 |
Mito Complexes, inhibition of c1 |
immediate |
EC50 |
0,105 µM |
van der Stel 2020, Figure 4 + Table 2 Tebby 2022, Figure 6 |
1, 3,4 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
EC25 |
0,016 µM |
Alimohammadi et al. 2023; Fig S4 |
5 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
10 µM |
Delp_2021, Figure 8ABC |
4 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
EC50 |
~ 0,1 µM |
ENVJMMONO(2020)22 van der Stel, Fig6AB / Annex 1.2 Tebby 2022, Fig4 |
2, 5 |
|
KE 177 |
LUHMES DoD3 |
ATP content |
DoD2-DoD3 (24h) |
EC25 |
10 µM |
Delp 2021, Fig S7+Fig4 |
|
|
KE 177 |
LUHMES DoD3 |
ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
7,8 µM |
ENVJMMONO(2020)22 van der Stel, Fig11 + Fig13 |
|
|
KE 177 |
LUHMES DoD3 |
ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0,0045 µM |
ENVJMMONO(2020)22 van der Stel, Fig13 |
|
|
KE 177 |
SH-SY5Y DoD7 |
mitochondrial membrane potential (via rhodamine123) |
DoD6-DoD7 (24h) |
EC25 |
0,316 µM |
Delp_2021 Figure 7 ENVJMMONO(2020)22 van der Stel Fig8 / Annex 1.6 |
3 |
|
KE 177 |
SH-SY5Y DoD7 |
ATP content |
DoD6-DoD7 (24h) |
EC50 |
> 10 µM |
ENVJMMONO(2020)22 van der Stel, Fig10 |
|
|
KE 177 |
SH-SY5Y DoD8 |
ATP content |
DoD3+DoD6-DoD8 (120h) |
EC50 |
1 µM |
ENVJMMONO(2020)22 van der Stel, Fig10 / Annex 1.16 |
1Permeabilized cells.
2EC estimated visually from graph.
3Likely same underlying data reprinted
4Only n=2
5Number of biological replicates unclear
Rotenone
KE 887: Five studies conducted in two cell lines (HepG2, LUHMES) measured inhibition of c1 immediately after treatment of cells with rotenone. Two studies in proliferating LUHMES cells were conducted only at a single, high concentration (10 and 100 µM). The few concentration-response experiments measured c1 inhibition at 10-30 nM for both LUHMES and HepG2 cells. Of note, all experiments were conducted in permeabilized cells. Some experiments consisted of only two biological experiments.
KE 177: Multiple studies measured the OCR in differentiating LUHMES immediately after treatment with rotenone. The lowest effective concentration was at 10 nM. When cells were treated for 24 h, OCR was affected at 100 – 500 nM. In the same exposure scenario, MMP was 24 nM, but ATP levels only decreased at 5-40 µM, when cells were cultured with glucose-containing medium. When fed with galactose, cells were more sensitive, with MMP and ATP affected at 0.2 nM and 0.01-6 nM, respectively. This corresponds to a shift of 200 to 1000-fold.
In differentiated SH-SY5Y cells, 24 h exposure to rotenone lead to a decrease in mitochondrial membrane potential at 40-100 nM as measured via rhodamine 123, but ATP content was not affected up to 10 µM, which was the highest tested concentration. A prolonged exposure to rotenone (120 h) however, affected the ATP content at 100 nM. To summarize, rotenone is potent in disturbing the mitochondrial membrane potential, but an effect on ATP and viability can only be observed upon prolonged exposure.
|
KE |
cell type |
assay |
exposure |
effect level |
effect conc |
reference |
notes |
|
KE 887 |
LUHMES (proliferating) |
Mito Complexes, inhibition of c1 |
immediate |
25% |
10 µM (single conc) |
Delp 2019, Figure 7, Suppl. Item 5 |
1 |
|
KE 887 |
LUHMES (proliferating) |
Mito Complexes, inhibition of c1 |
immediate |
25% |
100 µM (single conc) |
Delp 2021, Figure S8 |
1 |
|
KE 887 |
LUHMES (proliferating) |
Mito Complexes, inhibition of c1 |
Immediate |
EC25 (Tebby) EC50 (van der Stel) |
10 nM (Tebby) 0.0264 µM (van der Stel) |
Tebby 2022, Fig 4 + Table S2 ENVJMMONO(2020)22 van der Stel, Figure 4+5 |
1, 2, 3, 4 |
|
KE 887 |
HepG2 |
Mito Complexes, inhibition of c1 |
immediate |
EC25 |
10 nM |
Tebby 2022, Fig 6 + Table S4 |
1, 2, 4 |
|
KE 887 |
HepG2 |
Mito Complexes, inhibition of c1 |
immediate |
EC50 |
0.034 µM |
van der Stel 2020, Figure 4 + Table 2 |
1 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
10 µM |
Delp 2021, Figure 8 ABC |
|
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
10 µM |
ENVJMMONO(2020)22 van der Stel, Fig6AB / Annex 1.2 |
5 |
|
KE 177 |
LUHMES (intact) |
OCR |
immediate |
EC50 |
0.01 µM (basal respiration) |
Tebby 2022, Table S1 |
|
|
KE 177 |
LUHMES DoD3 |
OCR |
DoD2-DoD3 (24h) |
EC50 |
0.5 µM (basal respiration) 0.1 µM (maximal respiration) |
ENVJMMONO(2020)22 van der Stel, Fig6 C |
|
|
KE 177 |
LUHMES DoD3 |
MMP (via TMRE) (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
0.024 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
MMP (via TMRE) (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.0002 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
25.12 µM |
Delp 2019, Figure 3 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content |
DoD2-DoD3 (24h) |
EC25 |
5.011 µM |
Delp 2021, Figure 4 + Figure S7 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC50 |
40 µM |
ENVJMMONO(2020)22 van der Stel, Figure 13 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
> 1 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0.0056 µM = 5,6 nM |
Delp 2019, Figure 3 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC50 |
0.0048 µM = 4,8 nM |
ENVJMMONO(2020)22 van der Stel, Figure 13 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.00001 µM = 0,01 nM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
ATP production |
DoD2-DoD3 (24h) |
EC50 |
0.1 µM |
ENVJMMONO(2020)22 van der Stel, Fig6 C |
|
|
KE 177 |
LUHMES DoD3 |
ATP Production |
DoD2-DoD3 (24h) |
EC50 |
100 µM |
ENVJMMONO(2020)22 van der Stel, Fig 11 |
|
|
KE 177 |
SH-SY5Y DoD7 |
MMP (Rhodamine 123) |
DoD6-DoD7 (24h) |
EC25 |
0.040 µM |
Delp 2021, Figure 7 |
|
|
KE 177 |
SH-SY5Y DoD7 |
MMP (Rhodamine 123) |
DoD6-DoD7 (24h) |
EC50 |
0.1 µM |
ENVJMMONO(2020)22 van der Stel, Fig8 Annex 1.6 |
|
|
KE 177 |
SH-SY5Y DoD7 |
intracellular ATP content |
DoD6-DoD7 (24h) |
EC50 |
>10µM |
ENVJMMONO(2020)22 van der Stel, Fig 10 Annex 1.6 |
|
|
KE 177 |
SH-SY5Y DoD8 |
intracellular ATP content |
DoD3-DoD8 (120h) |
EC50 |
0.1µM |
ENVJMMONO(2020)22 van der Stel, Fig 10 Annex 1.6 |
1 Permeabilized cells.
2 EC estimated visually from graph.
3 Likely same underlying data reprinted
4 Only n=2
5 Number of biological replicates unclear
Fenpyroximate
KE 887: Multiple studies measured that 50 µM fenpyroximate inhibited c1 by abou 80% in proliferating LUHMES cells. The only concentration-response study was conducted in HepG2, which estimated an EC50 of 0.019 µM.
KE 177: In intact LUHMES exposed in glucose medium, OCR was reduced with 20 µM fenpyroximate (only 2 biological replicates of a single concentration). MMP, as measured via TMRE, was affected at 26 µM and ATP content at 13-15 µM. : In galactose-containing medium, MMP was affected at 0.2 nM, and ATP was affected at 0.7 nM. These values correspond to a shift of 10.000 fold.
Only one study investigated the effect of fenpyroximate on K177 in SH-SY5Y cels. MMP (measured via rhodamine123) was affected at 0.79 µM.
|
KE |
cell type |
assay |
exposure |
effect level |
effect conc |
reference |
notes |
|
KE 887 |
LUHMES (proliferating) |
complex inhibition (C1) |
immediate |
25% |
50 µM (single conc) |
Delp 2019, Supp Item 5 |
1 |
|
KE 887 |
LUHMES (proliferating) |
complex inhibition (C1) |
immediate |
25% |
50 µM (single conc) |
Delp 2021, Figure S8 |
1 |
|
KE 887 |
LUHMES (proliferating) |
complex inhibition (C1) |
immediate |
25% |
50 µM (single conc) |
Tebby 2022, Table S3 |
1 |
|
KE 887 |
HepG2 |
complex inhibition (C1) |
immediate |
EC50 |
0.019 µM |
van der Stel 2020, Figure 4 + Table 2 |
1 |
|
KE 177 |
LUHMES DoD3 |
OCR |
immediate |
25% |
20 µM |
Delp 2021, Fig 8A |
2 |
|
KE 177 |
LUHMES DoD3 |
MMP (TMRE) (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
26 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
MMP (TMRE) (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.0002 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content |
DoD2-DoD3 (24h) |
EC25 |
12.59 µM |
Delp 2021, FigS7 +Fig 4 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Glc) |
DoD2-DoD3 (24h) |
EC25 |
15 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content (+Gal) |
DoD2-DoD3 (24h) |
EC25 |
0.0007 µM |
Alimohammadi 2023, Fig 13 + S2 |
|
|
KE 177 |
SH-SY5Y DoD7 |
MMP (Rhodamine 123) |
DoD6-DoD7 (24h) |
EC25 |
0.79 µM |
Delp 2021, Fig 7 |
Pyrimidifen
KE 887: Four studies conducted in two cell lines (HepG2, LUHMES) measured inhibition of c1 immediately after treatment of cells with pyrimidifen. All three studies in LUHMES cells were conducted with proliferating LUHMES at a single, high concentration (50 µM), which inhibited OCR by about 80%. In HepG2, c1 was inhibited at low nanomolar concentrations (EC50 = 7 nM). Of note, all experiments were conducted in permeabilized cells.
KE 177: Only two studies measured KE2 in LUHMES cells: At a concentration of 12.5 uM, pyrimidifen reduced OCR by 75% in intact cells. Of note, this study consisted of only two biological replicates at a single chemical concentration. ATP levels were affected at 20 uM when cells were exposed to pyrimidifen for 24 h.
|
KE |
cell type |
assay |
exposure |
effect level |
effect conc |
reference |
notes |
|
KE 887 |
LUHMES (proliferating) |
complex inhibition (C1) |
immediate |
25% |
50µM (single conc) |
Delp 2019, Supp Item 5 |
1 |
|
KE 887 |
LUHMES (proliferating) |
complex inhibition (C1) |
immediate |
25% |
50µM (single conc) |
Delp 2021, Figure S8 |
1 |
|
KE 887 |
LUHMES (proliferating) |
complex inhibition (C1) |
immediate |
25% |
50µM (single conc) |
Tebby 2022, Table S3 |
1 |
|
KE 887 |
HepG2 |
complex inhibition (C1) |
immediate |
EC50 |
0.007 µM |
van der Stel 2020, Figure 4 + Table 2 |
1 |
|
KE 177 |
LUHMES |
OCR |
immediate |
25% |
12.5 µM |
Delp 2021, Figure 8ABC |
2 |
|
KE 177 |
LUHMES DoD3 |
intracellular ATP content |
DoD2-DoD3 (24h) |
EC25 |
20 µM |
Delp 2021, Fig S7 + 4 |
|
|
KE 177 |
SH-SY5Y |
MMP (Rhodamine 123) |
DoD6-DoD7 (24h) |
EC25 |
0.79 µM |
Delp 2021, Figure 7 |
1 Permeabilized cells.
2 Only n=2
Tebufenpyrad
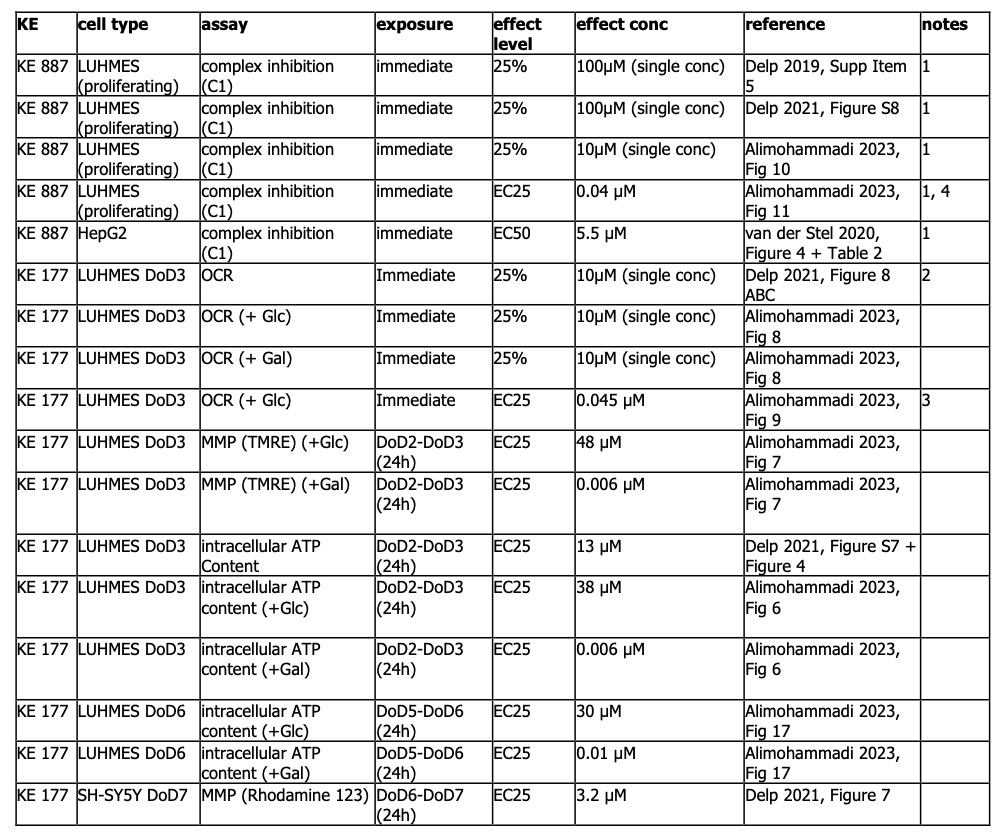
KE 887: Five studies conducted in two cell lines (HepG2, LUHMES) measured inhibition of c1 immediately after treatment of cells with tebufenpyrad. Three studies in proliferating LUHMES cells were conducted only at a single, high concentration (10 and 100 µM). Mitochondrial respiration was inhibited by 75% or more. For concentration-response, only one biological replicate was performed in LUHMES cells (EC25 = 40 nM). In HepG2 cells, the EC50 was considerably higher, at 5.5 uM. Of note, all experiments were conducted in permeabilized cells.
KE 177: There is strong evidence for tebufenpyrad decreasing OCR also in unpermeabilized (intact) LUHMES cells. This decrease was observed in both, glucose medium and galactose medium conditions. Only one study performed the experiment in concentration-response (EC25 = 45 nM), but the number of biological and technical replicates was unclear.
Glucose: In glucose medium, MMP was affected at 48 uM, and ATP content between 13 – 38 uM. Galactose: In galactose medium, MMP was affected at 6 nM and ATP content between 6-10 nM.
Only one study measured the effect of tebufenpyrad on KE 177 in SH-SY5Y cells. MMP was affected at 3.2 uM
1 Permeabilized cells.
2 Only n=2
3 Number of biological replicates unclear
4only one biological replicate
-Not endorsed
Uncertainties and Inconsistencies
Some studies suggest that rotenone may have effects other than CI inhibition, and it has been claimed that rotenone induces microtubule disruption, rather than ETC CI inhibition (Feng, 2006; Ren et al., 2005). Some studies suggested that there was no evidence for significant change in mitochondrial CI function in PD patients' brains (Jenner et al., 1992). It is still unclear whether the site of superoxide production in CI inhibited mitochondria is CI itself or not (Singer and Ramsay, 1994).
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
|
Uncertainty |
Impact |
Reason |
|
KE 887 measured in permeabilised cells |
|
Permeabilisation provides direct access for the tested compounds and substrates to the mitochondria and respiratory chain components. The physicochemical properties of the tested compound may reduce its ability to permeate the plasma membrane of intact cells, thus reducing or preventing its uptake. |
|
Lack of data for some endpoints in galactose condition |
High |
In vitro cell models in general are characterized by an unphysiological reliance on glycolysis. In the presence of glucose any KE is influenced by the contribution of oxidative phosphorylation in addition to glycolisis to meet the cellular need for ATP. Thus, the KEs are influenced by the glycolisis rate. Glucose concentrations in culture medium higher than the physiological level enhances cellular resistance to mitochondrial dysfunction. Application of galactose instead of glucose in the medium allows a shift towards mitochondrial ATP generation. Even under these conditions, glycolysis significantly contributes to ATP production. |
|
Use of HepG2 concentration response curves related to the measurement of oxygen consumption upon inhibition of cIII as a surrogate to represent inhibition of cIII in LUHMES cells, due to the lack of concentration response data for LUHMES cells
|
Low |
It is assumed that since the exposure is acute and in permeabilized cells, the test chemical would have immediate access to the mitochondria. Other mechanisms such as transport into the cells, ADME considerations or an indirect effect via other signaling pathways were considered negligible under these assay conditions. It should be noted that OCR was measured in the presence of glucose, which introduces an influence from the glycolitic rate. This factor may differ between hepatocytes and neurons. |
|
KE 177 - no concentration-response data for OCR in LUHMES
|
High |
Increase the uncertainty in the concordance concentration response relationship across the KEs |
|
Methodological limits |
Medium-low |
• For some assays and chemicals, only two biological replicates were performed (instead of 3), therefore results should be considered with caution. • Different assays have a different effect concentration (i.e. EC25 and EC50). Occasionally, also the same assay can have different effect levels depending on the publication, which reduces overall comparability. However, in most cases the EC25 and EC50 are within a factor of 3 of each other, thus limiting the uncertainty. |
- Not endorsed
Known modulating factors
Quantitative Understanding of the Linkage
Based on the available data, the threshold effect seen in brain mitochondria indicates that modest CI inhibition (~ 25-50% decrease in activity) may not directly impact ATP levels or Δψm. Indeed, low levels of CI inhibition produces an oxidative stress without any significant changes in mitochondrial respiration (Betarbet et al., 2000; Greenamyre et al., 2001) or causes not significant changes in ATP levels (Sherer et al., 2003). In particular, in rotenone-infused animals (2.0 mg/kg per day for 2 days), [3H] dihydrorotenone binding to CI in brain was reduced by about 73%. Based on this degree of binding inhibition, the rotenone concentration in brain was estimated to be between 20−30 nM. Complexes II and IV were unchanged by rotenone infusion (Betarbet et al., 2000).
However, such defects have long-term deleterious effects. It is well documented that that there is a site of electron leak upstream of the rotenone binding site in CI (i.e., on the ‘NADH side’ of the complex) (Hensley et al., 1998) leading to the superoxide (O2-) and followed up by H2O2 production by CI (Greenamyre et al., 2001). The relative role of each ETC complex in forming superoxide differs by tissue; however CI is a major source of O2- in the brain (Halliwell, 2007).
Thus, a low inhibition of CI activity that is insufficient to affect cell respiration may lead to mitochondrial damage and chronic up-regulation of ROS production. Therefore, it is suggested that rotenone that binds to CI with an affinity of 10-20 nM induces toxicity not by bioenergetics effects but rather via accumulative oxidative stress. Sustained oxidative stress leads to decrease levels of reduced glutathione; activation of superoxide dismutase (SOD) (scavenger of O2-), catalase and indeed, treatments with antioxidants reduce the oxidative stress-induced damage. Such data are abounded in the existing literature based both on in vivo and in vitro studies and a few examples are described in the Empirical support for linkage.
The selective CI defects (other complexes were unaffected) (Schapira et al., 1990a) and induced mitochondrial damage followed by oxidative stress is also described in PD patients brains as documented by: (a) reduced glutathione levels (Jenner at al., 1992); (b) increased content of 8-oxo-deoxyguanine, a marker of oxidatively damaged nucleic acids (Alam et al., 1997; Mecocci et al., 1993); (c) increased level of malondialdehyde (marker of lipid peroxidation) (Navarro et al., 2009); (d) increased cholesterol lipid hydroperoxide (Dexter et al., 1994); (e) increased protein oxidation measured e.g. by elevated levels of methionine sulfoxide formation or protein carbonyl content (Alam et al., 1997). These studies in human brain present a semiquantitataive evaluation of the oxidative stress, as there is no data showing KER between the various degrees of CI inhibition and mitochondrial damage (ROS production) and the parameters described above. However, these studies clearly confirmed that oxidative stress in PD patient brain is increased as shown by the measured biomarkers (Sanders and Greenamyre, 2013).
In in vitro and in in vivo animal studies there are some data showing the quantitative relationship between the oxidative stress produced by inhibition of CI and mitochondrial damage measured by the same assays, as described in human studies, and a few examples of such experiments are discussed below. The quantitative evaluation of the causative relationship between the CI inhibition (KE up) induced by rotenone (4 hr exposure) and mitochondrial dysfunction (KE down) measured in human-chimpanzee isolated mitochondria (xenomitochondrial cybrids; HXC) by a decreased cell respiration and Δψm, increased ROS production and lipid peroxidation showed linear, time- and concentration-dependent effects (below Fig.1 from Barrientos and Moraes, 1999).

Fig.1. A dose- and time-dependent effect of CI inhibition by rotenone on (A) reactive oxygen species production (ROS), (B) Lipid peroxidation and (C) mitochondrial membrane potential (Δψm) studied in the human osteosarcoma-derived cell line (143B) or using a genetic model (40% CI inhibited in HXC lines) (for further information see Barrientos and Moraes, 1999).
- The endogenous respiration was inhibited in a dose-dependent manner but showed different inhibition kinetics. Only when CI was inhibited by 35-40% (< 5 nM rotenone), cell respiration started decreasing (a threshold for inhibition for cell respiration triggered by rotenone). Between 40 and 60% of CI inhibition (5-10 nM), cell respiration decreased linearly until 30% of the normal rate. Increasing concentrations of rotenone produced further but slower decrease in CI activity and cell respiration. 100% CI inhibition was achieved with 100 nM rotenone but the cells still maintained a cell respiration rate (through complex II), approximately 20% and the rate of ROS production increased by a maximum of 20-25% (4 hr treatment). ROS production was saturated at 100 nM rotenone but an initial effect was observed already at 1-5 nM (Barrientos and Moraes, 1999). Inhibition of CI activity triggered decrease of cell respiration by different concentrations of rotenone and resulted in mitochondrial damage measured not only by ROS production, but also by lipid peroxidation and decreased Δψm. Inhibition of CI by 25, 50, 75 and 100 % decreased cell respiration by 5, 20, 53, 81 %, increased ROS production by 48, 81, 157, 216%, increased lipid peroxidation by 8, 27, 45, 55 % and decreased Δψm by 6, 13, 20, and 37% respectively (approximately).
Similar studies were also performed using different types of neuronal cells.
- Hoglinger and colleagues, by using DA neurones derived from the rat (embryonic day 15.5) ventral mesencephalon, showed that CI inhibition by rotenone at 30 nM, (or MPP+ 3 µM) for 24 hr decreased ATP levels (by > 80%) within the first 6 hr, and neuronal cell death within 24 hr. When residual ATP levels remained above 20%, there was no or little neuronal loss, suggesting that 20% of normal ATP level was the minimum compatible with neuronal survival. Rotenone (and MPP+) increased ROS (≥ 40% over control levels) already at low concentrations that were subtoxic or only moderately toxic (i.e., 10-30 nM for rotenone, 10-30 µM for MPP+) (Fig. 2) (Hoglinger et al., 2003).

Fig. 2. ATP levels, ROS production and neuronal surviving cells in mesencephalic cultures treated with CI inhibitors (rotenone and MPP+) (from Hoglinger et al., 2003, Fig. 4a-b)
- Shamoto-Nagai and colleagues showed that 25 or 50 nM rotenone decreased ATP levels over time. In particular, the intracellular ATP level was reduced to 18.0% and 19.6% of control after 44 hr of treatment with 25 and 50 nM of rotenone, respectively, and thereafter the decreased level was sustained (Fig. 3, left) (Shamoto-Nagai et al., 2003). Also, The production of ROS-RNS increased 6 hr after the rotenone treatment, and the increase was about 1.5-fold of the basal value. With treatment with the higher (50 nM) concentration of rotenone, DCF production level was restored to the basal level after 48 hr, whereas, at the lower concentration (25 nM), DCF production increased again at 48 hr and then declined to the basal value after 90 hr (Fig. 3, right) (Shamoto-Nagai et al., 2003).

Fig. 3. Effect of rotenone on ATP level (left) and on ROS and RNS production (right) in SH-SY5Y cells. SH-SY5Y cells were treated with 25 nM (circles) or 50 nM (triangles) of rotenone. * indicates significant difference from control (P < .05) (from Shamoto-Nagai et al., 2003, Figs. 2, 3)
- Human neuroblastoma cell line (SK-N-MC) exposed to 5 nM rotenone chronically, for 4 weeks caused reduction in GSH by 44%, GSSG by 40%. These effects were not observed after two weeks of exposure. Total cellular GSH levels were reduced after 4 weeks of exposure by 50% (Sherer et al., 2002). Similarly, in the same study, 1-2 weeks of treatment did not alter protein carbonyl levels (oxidative protein damage) but exposure for 3-4 weeks caused a large increase in carbonyls in the insoluble fraction by approximately 223% of control. Systemic in vivo rotenone infusion (up to 5 weeks, 3.0 mg/kg/day) modestly elevated soluble protein carbonyls in the rat cortex by approximately 19%, in the striatum by 27% and the largest elevation occurred in the DA neurons of midbrain, around 41% (no effect in cerebellum or hippocampus) (Sherer et al., 2003).
The prolonged treatment with rotenone (3-4 weeks, not 1-2 weeks) caused also a marked increase in 8-oxo-dG immune-reactivity (i.e., oxidative DNA damage) and redistribution of cytochrome c (Sherer et al., 2002).
- The same group showed that exposure of SK-N-MC cells for 6-8 hr to low concentrations of rotenone (100 pM, 1 nM, 10 nM and 100 nM) produced a concentration-dependent decrease in ATP levels by 0, 2.5, 10, and 32.2 % respectively (Sherer et al., 2003).
- The oxidative stress (mitochondrial damage) induced by rotenone exposure was confirmed in ex-vivo studies using brain sections at the level of the substantia nigra that were treated with 50 nM rotenone over 1 week. A significant increase of protein carbonyls (indicative of oxidative damage to proteins; biomarkers of oxidative stress) was observed (~ 25%) when compared to the untreated slices. Exposure to 100 μM α-tocopherol, antioxidant (vitamin E) significantly protected the neurons from the oxidative damage induced by 50 nM rotenone over 1 week (~ 25%), as shown by lower protein carbonyl levels (~ 3%), with very similar effects observed with 20 nM rotenone over 2 weeks (Testa et al., 2005).
The same assays for mitochondrial dysfunction evaluation after exposure to rotenone, MPTP or other chemicals were used through a range of different studies (Sherer et al., 2003, Betarbet et al., 2000) and the role of CI inhibition in PD is discussed in many published reviews (Sanders and Greenamyre, 2013, Greenamyre et al., 2001, Schapira et al., 1990a and 1990b).
Summing up, it is well documented in human PD brain studies as well as in vivo and in vitro existing data that CI inhibition induces mitochondrial dysfunction as shown by measuring the decreased cellular respiration and induced oxidative damage to protein, lipids and nucleic acids, as well as compromised function of anitioxidant defense mechanisms (e.g. decreased levels of reduced glutathione). As discussed above, oxidative damage is largely reversed by antioxidants treatments. These data are largely semi quantitative only, as the full dose- and time response curves are available. They indicate that low levels of CI inhibition for long periods of time (4-5 weeks) mostly increase ROS production, having negative effects on DA neurons in SNpc, which seem to be affected more than other neuronal cell types in other brain structures (reviews e.g. by Sanders and Greenamyre, 2013; Greenamyre et al., 2001, Schapira et al., 1990a and 1990b etc.).
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
The quantitative understanding of the KERs was gained by modelling the KERs within the qAOP framework and methods that were developed in Tebby et al., (2022). Data and uncertainties are reported in "quantitative understanding" section of AOP 3 (cI inhibitor).
- Not endorsed
Response-response Relationship
- Revision of AOP3 (Project: NP/EFSA/PREV/2024/02):
An overview of these data across AOPs and KEs, summarising the percentage effect on each KE, is presented in the “Evidence assessment” section of AOP 3 (cI inhibitor).
- Not endorsed
Time-scale
Known Feedforward/Feedback loops influencing this KER
Domain of Applicability
Mitochondrial CI in eukaryotes has highly conserved subunit composition based on protein databases (Cardol, 2011). The characterization of induced mitochondrial dysfunction phenotypes in zebrafish was studied in the presence of CI and CII inhibitors (Pinho et al., 2013). Exposure of Caenorhabditis elegans (C. elegans) to rotenone, reduced bioluminescence (an assay for mitochondrial dysfunction) after both relatively short (2 hr) and longer exposures (24 hr) to a range of concentrations. A sharp decline in bioluminescence (maximal inhibition) relative to controls occurred at the lowest rotenone concentration of 2.5 μM. This decline in bioluminescence was consistent with reduced cellular ATP (Lagido et al., 2015). The results obtained from C. elegans exposed to rotenone suggested that chronic exposure to low concentration (2 or 4 μM) caused mitochondrial damage through persistent suppression of mitochondrial biogenesis and mitochondrial gene expression leading to mitochondrial dysfunction that contributed to DA neuron degeneration (Zhou et al., 2013).
Drosophila melanogaster has been proven suitable to study signaling pathways implicated in the regulation of mitochondrial function and integrity, such as the PINK1/parkin pathway (controlling mitochondrial integrity and maintenance), DJ-1 and Omi/HtrA2 genes (associated with the regulation of mitochondrial functionality). Notably, PINK1, PARKIN, and DJ-1 genes are associated with recessive forms of PD (Guo, 2012). Drosophila flies lacking DJ-1 result to be viable, but show an increased sensitivity to oxidative stress induced upon rotenone or Paraquat (an herbicide inducer of CI-dependent ROS) feeding (Menzies et al. 2005; Meulener et al. 2005; Meulener et al. 2006). Moreover, it has been reported in Drosophila that inhibition of CI by mean of sublethal chronic exposure to rotenone (<750 μM) via the feeding medium caused a selective loss of DA neurons in all of the brain regions and locomotor impairments, while L-dopa (3,4-dihydroxy-L-phenylalanine) rescued the behavioral deficits (but not neuronal death) (Coulom and Birman, 2004). MPTP causes Parkinsonism in primates including humans. However, rodents (rats) are much less susceptible to MPTP+ but are fully susceptible to MPP+ (due to the differences in toxicokineticks). In all species, CI inhibition leads to mitochondrial dysfunction. Mitochondrial dysfunction is an universal event occurring in cells of any species (Farooqui and Farooqui, 2012).
References
Alam ZI, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997; 69(3):1196–203.
Alimohammadi Mahshid, Birthe Meyburg, Anna-Katharina Ückert, Anna-Katharina Holzer,
Marcel Leist, 2023. EFSA Pilot Project on New Approach Methodologies (NAMs) for
Tebufenpyrad Risk Assessment. Part 2. Hazard characterisation and identification of the
Reference Point. EFSA supporting publication 2023:EN-7794. 56 pp.
doi:10.2903/sp.efsa.2023.EN-7794
Ambrosi G, Ghezzi C, Sepe S, Milanese C, Payan-Gomez C, Bombardieri CR, Armentero MT, Zangaglia R, Pacchetti C, Mastroberardino PG, Blandini F. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson's disease. Biochim Biophys Acta. 2014 Sep;1842(9):1385-94.
Barrientos A, and Moraes CT. 1999. Titrating the Effects of Mitochondrial Complex I Impairment in the Cell Physiology. 274(23):16188–16197.
Bennekou, S. H., van der Stel, W., Carta, G., Eakins, J., Delp, J., Forsby, A., Kamp, H., Gardner, I., Zdradil, B., Pastor, M., Gomes, J. C., White, A., Steger-Hartmann, T., Danen, E. H. J., Leist, M., Walker, P., Jennings, P., & van de Water, B. (2020).ENV/JM/MONO(2020)23 Case study on the use of integrated approaches to testing and assessment for mitochondrial complex-iii-mediated neurotoxicity of azoxystrobin - read-across to other strobilurins: Series on testing and assessment no. 327. Organisation for Economic Co-operation and Development.
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 3:1301-1306.
Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999 Oct; 79(4):1127-55.
Braun RJ. 2012. Mitochondrion-mediated cell death: dissecting yeast apoptosis for a better understanding of neurodegeneration. Front Oncol 2:182.
Brown GC, and Borutaite V. 2004. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols, Biochimica et Biophysica Acta (BBA) – Bioenergetics 1658, 1–2.
Cardol P. 2011. Mitochondrial NADH:ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases.". Biochim Biophys Acta 1807 (11): 1390–7.
Chen Y, Zhang DQ, Liao Z, Wang B, Gong S, Wang C, Zhang MZ, Wang GH, Cai H, Liao FF, Xu JP 2015. Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson's disease. Mol Neurodegener. 2;10(1):4.
Cheng B, Guo Y, Li C, Ji B, Pan Y, Chen J, Bai B. .Edaravone protected PC12 cells against MPP(+)-cytoxicity via inhibiting oxidative stress and up-regulating heme oxygenase-1 expression. J Neurol Sci. 2014 Aug 15;343(1-2):115-9.
Chiu CC, Yeh TH, Lai SC, Wu-Chou YH, Chen CH, Mochly-Rosen D, Huang YC, Chen YJ, Chen CL, Chang YM, Wang HL, Lu CS. 2015. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp Neurol. 263:244-53.
Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. J Neurosci. 2004 Dec 1;24(48):10993-8.
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. 2012. Mitochondrial importance in Alzheimer's, Huntington's and Parkinson's diseases. Adv Exp Med Biol 724:205221.
Cozzolino M, Ferri A, Valle C, Carri MT. 2013. Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci 55:44–49.
Delp J, Cediel-Ulloa A, Suciu I, Kranaster P, van Vugt-Lussenburg BM, Munic Kos V, van der Stel W, Carta G, Bennekou SH, Jennings P, van de Water B, Forsby A, Leist M. Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors. Arch Toxicol. 2021 Feb;95(2):591-615. doi: 10.1007/s00204-020-02970-5. Epub 2021 Jan 29. PMID: 33512557; PMCID: PMC7870626.
Delp J, Funke M, Rudolf F, Cediel A, Bennekou SH, van der Stel W, Carta G, Jennings P, Toma C, Gardner I, van de Water B, Forsby A, Leist M. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch Toxicol. 2019 Jun;93(6):1585-1608. doi: 10.1007/s00204-019-02473-y. Epub 2019 Jun 12. PMID: 31190196.
Dexter DT, et al. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994; 9(1):92–7.
Dias V, Junn E. and Mouradian MM. 2013. “The role of oxidative stress in parkinson’s disease,” Journal of Parkinson’s Disease, 3(4)461–491.
EFSA Panel on Plant Protection Products and their residues (PPR); Ockleford C, Adriaanse P, Berny P, Brock T, Duquesne S, Grilli S, Hernandez-Jerez AF, Bennekou SH, Klein M, Kuhl T, Laskowski R, Machera K, Pelkonen O, Pieper S, Smith R, Stemmer M, Sundh I, Teodorovic I, Tiktak A, Topping CJ, Wolterink G, Angeli K, Fritsche E, Hernandez-Jerez AF, Leist M, Mantovani A, Menendez P, Pelkonen O, Price A, Viviani B, Chiusolo A, Ruffo F, Terron A, Bennekou SH. Investigation into experimental toxicological properties of plant protection products having a potential link to Parkinson's disease and childhood leukaemia. EFSA J. 2017 Mar 16;15(3):e04691. doi: 10.2903/j.efsa.2017.4691. PMID: 32625422; PMCID: PMC7233269.
Farooqui T. and Farooqui, AA. 2012. Oxidative stress in Vertebrates and Invertebrate: molecular aspects of cell signalling. Wiley-Blackwell, Chapter 27:377- 385
Feng J. Mictrotubule. A common target for parkin and Parkinson's disease toxins. Neuroscientist 2006, 12.469-76.
Filomeni G, Graziani I, de Zio D, Dini L, Centonze D., Rotilio G, Ciriolo MR. 2012. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging. 33:767–785.
Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, Diederich N, Buttini M, Kodama A, Satagopam VP,
Eifes S, Del Sol A, Schneider R, Kitano H, Balling R. 2014. Integrating pathways of Parkinson's disease in a molecular interaction map. Mol Neurobiol.49(1):88-102.
Graier WF, Frieden M, Malli R. 2007. Mitochondria and Ca2+ signaling: old guests, new functions. Pflugers Arch 455:375–396.
Greenamyre JT, Sherer TB, Betarbet R, and Panov AV. 2001. Critical Review Complex I and Parkinson’s Disease. Life. 52:135–141.
Guo M. Drosophila as a model to study mitochondrial dysfunction in Parkinson's disease. Cold Spring Harb Perspect Med. 2012 Nov 1;2(11). pii: a009944. Halliwell, BaG; JMC. Free Radicals in Biology and Medicine. 4. Oxford University Press; 2007.
Hartman P, Ponder R, Lo HH, Ishii N. Mitochondrial oxidative stress can lead to nuclear hypermutability. Mech Ageing Dev. 2004 Jun;125(6):417-20.
Hensley, K., Pye, Q. N., Maidt, M. L., Stewart, C. A., Robinson, K. A., Jaffrey, F., and Floyd, R. A. (1998) Interaction of alpha-phenyl -N-tert-butyl nitrone and alternative electron acceptor s with complex I indicates a substrate reduction site upstream from the rotenone binding site. J. Neurochem. 71, 2549–2557.
Höglinger GU, Carrard G, Michel PP, Medja F, Lombès A, Ruberg M, Friguet B, Hirsch EC. 2003. Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J. Neurochem. 86, 1297–1307.
Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group. Ann Neurol. 1992;32 Suppl:S82-7
Lagido C., McLaggan D., and Glover L.A.. A . Screenable In Vivo Assay for Mitochondrial Modulators Using Transgenic Bioluminescent Caenorhabditis elegans. J Vis Exp. 2015; (104): 53083.
Lee DH, Kim CS, Lee YJ. Astaxanthin protects against MPTP/MPP+-induced mitochondrial dysfunction and ROS production in vivo and in vitro. Food Chem Toxicol. 2011 Jan;49(1):271-80.
Liu W, Kong S, Xie Q, Su J, Li W, Guo H, Li S, Feng X, Su Z, Xu Y, Lai X. Protective effects of apigenin against 1-methyl-4-phenylpyridinium ion induced neurotoxicity in PC12 cells. Int J Mol Med. 2015, 35(3):739-46.
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD. 1992. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain. 115 ( Pt 2):333-42.
Martin LJ. 2011. Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43:569–579.
Mecocci P, et al. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993; 34(4):609–16.
Menzies FM, Yenisetti SC, Min KT. 2005. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr Biol 15: 1578–1582.
Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM. 2005. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr Biol 15: 1572–1577.
Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM. 2006. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci 103: 12517–12522.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. 1989. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Commun. 163(3):1450-5
Nataraj J, Manivasagam T, Justin Thenmozhi A, Essa MM 2015. Lutein protects dopaminergic neurons against MPTP-induced apoptotic death and motor dysfunction by ameliorating mitochondrial disruption and oxidative stress. Nutr Neurosci. 2015 Mar 2. [Epub ahead of print].
Navarro A, et al. Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic Biol Med. 2009; 46(12):1574–80.
Ojha S, Javed H, Azimullah S, Abul Khair SB, Haque ME Neuroprotective potential of ferulic acid in the rotenone model of Parkinson's disease. Drug Des Devel Ther. 2015 Oct 7;9:5499-510.
Parker WD Jr, Boyson SJ, Parks JK. 1989. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol.26(6):719-23.
Piert M, Koeppe RA, Giordani B, Minoshima S, Kuhl DE. Determination of regional rate constants from dynamic FDG-PET studies in Parkinson's disease. J Nucl Med. 1996 Jul;37(7):1115-22.
Pinho BR, Santos MM, Fonseca-Silva A, Valentão P, Andrade PB, Oliveira JM. How mitochondrial dysfunction affects zebrafish development and cardiovascular function: an in vivo model for testing mitochondria-targeted drugs. Br J Pharmacol. 2013 Jul;169(5):1072-90.
Rango M, Bonifati C, and Bresolin N. 2006. Parkinson’s disease and brain mitochondrial dysfunction: a functional phosphorus magnetic resonance spectroscopy study.Journal of Cerebral Blood Flow & Metabolism. 26(2)283–290.
Ren Y. et al., 2005. Selectivwe vulnerabity of dopaminergic neurons to microtubule depolymerisation. J. Bio. Chem. 280:434105-12.
Ramsay RR, Singer TP. 1992. Relation of superoxide generation and lipid peroxidation to the inhibition of NADH-Q oxidoreductase by rotenone, piericidin A, and MPP+. Biochem Biophys Res Commun. 189(1):47-52.
Sanders LH, and Greenamyre JT. 2013. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic Biol Med.62:111-20.
Sanders LH, McCoy J, Hu X, Mastroberardino PG, Dickinson BC, Chang CJ, Chu CT, Van Houten B, Greenamyre JT. 2014. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiol Dis. 70:214-23.
Saravanan KS, Sindhu KM, Senthilkumar KS, Mohanakumar KP. 2006. L-deprenyl protects against rotenone-induced, oxidative stress-mediated dopaminergic neurodegeneration in rats. Neurochem Int.49(1):28-40.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, and Marsden CD. 1989. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1,1269.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. 1990a. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 54(3):823-7.
Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD. 1990b. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson's disease. J Neurochem. 55(6):2142-5.
Schapira AH. 1994. Evidence for mitochondrial dysfunction in Parkinson's disease--a critical appraisal. Mov Disord. 9(2):125-38.
Shamoto-Nagai M, Maruyama W, Kato Y, Isobe K, Tanaka M, Naoi M, Osawa T. 2003. An inhibitor of mitochondrial complex I, rotenone, inactivates proteasome by oxidative modification and induces aggregation of oxidized proteins in SH-SY5Y cells. J Neurosci Res. 74:589–97.
Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. 2002. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci. 22(16):7006-15.
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, et al. 2003. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 23:10756–64.
Singer TP, Ramsay RR.The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochim Biophys Acta. 1994 Aug 30;1187(2):198-202.
Takeshige K 1994. Superoxide formation and lipid peroxidation by the mitochondrial electron-transfer chain. Rinsho Shinkeigaku. 34(12):1269-71.
Tebby C, Gao W, Delp J, Carta G, van der Stel W, Leist M, Jennings P, van de Water B, Bois FY. A quantitative AOP of mitochondrial toxicity based on data from three cell lines. Toxicol In Vitro. 2022 Jun;81:105345. doi: 10.1016/j.tiv.2022.105345. Epub 2022 Mar 10. PMID: 35278637.
Testa CM, Sherer TB, Greenamyre JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res. 2005; 134(1):109–18.
Thomas B, Banerjee R, Starkova NN, Zhang SF, Calingasan NY, Yang L, Wille E, Lorenzo BJ, Ho DJ, Beal MF, Starkov A. Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson's disease. Antioxid Redox Signal. 2012 May 1;16(9):855-68.
Tseng YT, Chang FR, Lo YC2. 2014. The Chinese herbal formula Liuwei dihuang protects dopaminergic neurons against Parkinson's toxin through enhancing antioxidative defense and preventing apoptotic death. Phytomedicine. 21(5):724-33.
van der Stel W, Carta G, Eakins J, Darici S, Delp J, Forsby A, Bennekou SH, Gardner I, Leist M, Danen EHJ, Walker P, van de Water B, Jennings P. Correction to: Multiparametric assessment of mitochondrial respiratory inhibition in HepG2 and RPTEC/TERT1 cells using a panel of mitochondrial targeting agrochemicals. Arch Toxicol. 2020 Aug;94(8):2731-2732. doi: 10.1007/s00204-020-02849-5. Erratum for: Arch Toxicol. 2020 Aug;94(8):2707-2729. doi: 10.1007/s00204-020-02792-5. PMID: 32720191; PMCID: PMC7645484.
van der Stel, Wanda; Bennekou, Susanne Hougaard; Carta, Giada; Eakins, Julie; Delp, Johannes; Forsby, Anna; Kamp, Hennicke; Gardner, Ian; Zdradil, Barbara; Pastor, Manual (2020) ENV/JM/MONO(2020)22 CASE STUDY ON THE USE OF INTEGRATED APPROACHES TO TESTING AND ASSESSMENT FOR IDENTIFICATION AND CHARACTERISATION OF PARKINSONIAN HAZARD LIABILITY OF DEGUELIN BY AN AOP-BASED TESTING AND READ ACROSS APPROACH Series on Testing and Assessment No. 326
van der Stel W, Yang H, Vrijenhoek NG, Schimming JP, Callegaro G, Carta G, Darici S, Delp J, Forsby A, White A, le Dévédec S, Leist M, Jennings P, Beltman JB, van de Water B, Danen EHJ. Mapping the cellular response to electron transport chain inhibitors reveals selective signaling networks triggered by mitochondrial perturbation. Arch Toxicol. 2022 Jan;96(1):259-285. doi: 10.1007/s00204-021-03160-7. Epub 2021 Oct 13. PMID: 34642769; PMCID: PMC8748354.
Wang S, He H, Chen L, Zhang W, Zhang X, Chen J. Protective effects of salidroside in the MPTP/MPP(+)-induced model of Parkinson's disease through ROS-NO-related mitochondrion pathway. Mol Neurobiol. 2015, 51(2):718-28.
Wang H, Liu J, Gao G, Wu X, Wang X, Yang H. Protection effect of piperine and piperlonguminine from Piper longum L. alkaloids against rotenone-induced neuronal injury. Brain Res. 2015 Jul 29. pii: S0006-8993(15)00558-2. doi: 10.1016/j.brainres.2015.07.029. [Epub ahead of print].
Wu RM, Mohanakumar KP, Murphy DL, Chiueh CC. 1994. Antioxidant mechanism and protection of nigral neurons against MPP+ toxicity by deprenyl (selegiline). Ann N Y Acad Sci. 17;738:214-21.
Zhou S, Wang Z, Klaunig JE. Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. Int J Biochem Mol Biol. 2013 Dec 15;4(4):191-200. eCollection 2013.
Zhu J, and Chu CTT. 2010. Mitochondrial dysfunction in Parkinson’s disease. Journal of Alzheimer’s Disease, 20(2):S325–S334.