
This AOP is licensed under the BY-SA license. This license allows reusers to distribute, remix, adapt, and build upon the material in any medium or format, so long as attribution is given to the creator. The license allows for commercial use. If you remix, adapt, or build upon the material, you must license the modified material under identical terms.
AOP: 626
Title
Increased 11β-Hydroxysteroid dehydrogenase type 1 activity leading to MASLD progression via insulin resistance-associated endoplasmic reticulum stress
Short name
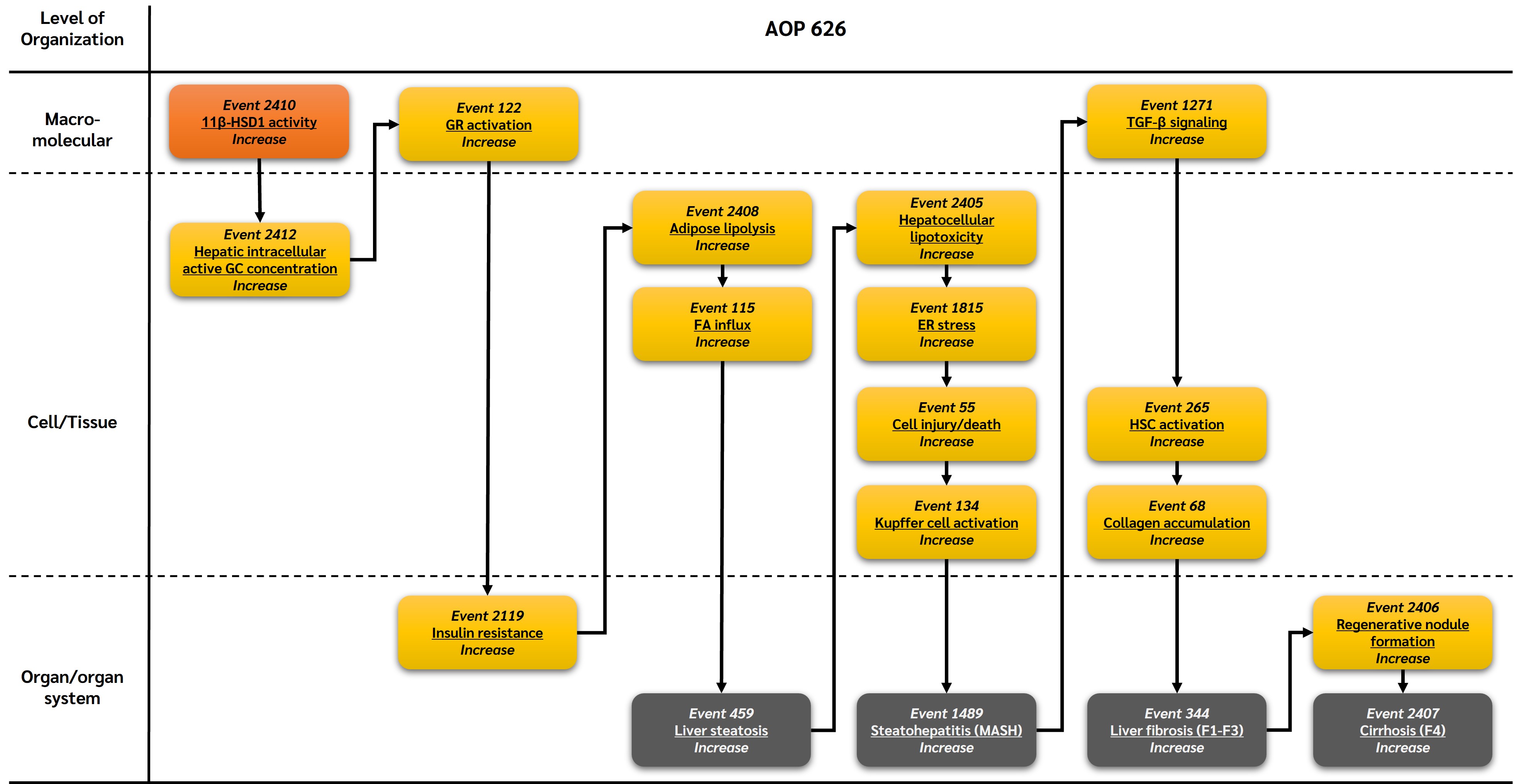
Graphical Representation
Additional AOP Exploration Options
Click links below to explore AOP 626, Increased 11β-Hydroxysteroid dehydrogenase type 1 activity leading to MASLD progression via insulin resistance-associated endoplasmic reticulum stress in tools offered by third parties.
Point of Contact
Contributors
- You Song
Coaches
- Shihori Tanabe
OECD Information Table
| OECD Project # | OECD Status | Reviewer's Reports | Journal-format Article | OECD iLibrary Published Version |
|---|---|---|---|---|
| Under Development |
This AOP was last modified on February 24, 2026 09:05
Revision dates for related pages
| Page | Revision Date/Time |
|---|---|
| Increase, 11β-Hydroxysteroid dehydrogenase type 1 activity | February 18, 2026 08:38 |
| Increase, Hepatic intracellular active glucocorticoids | February 26, 2026 06:48 |
| Increase, Glucocorticoid receptor activation | February 12, 2026 07:24 |
| Increase, Insulin resistance | February 11, 2026 05:50 |
| Increase, Adipose lipolysis | February 10, 2026 05:00 |
| Increased, Fatty acid influx | February 24, 2026 08:56 |
| Increase, Liver steatosis | February 11, 2026 05:41 |
| Increase, Hepatocellular lipotoxicity | February 10, 2026 04:40 |
| Increase, Endoplasmic reticulum stress | February 11, 2026 06:00 |
| Increase, Cell injury/death | May 27, 2024 07:23 |
| Increase, Kupffer cell activation | February 11, 2026 05:16 |
| Increase, Steatohepatitis | February 24, 2026 09:13 |
| Increase, Transforming growth factor-beta signaling | February 11, 2026 05:39 |
| Increase, Hepatic stellate cell activation | February 11, 2026 07:04 |
| Increase, Collagen accumulation | February 11, 2026 06:58 |
| Increase, Liver fibrosis | February 11, 2026 05:35 |
| Increase, Regenerative nodule formation | February 10, 2026 06:47 |
| Increase, Cirrhosis | February 11, 2026 07:34 |
| Increase, 11β-HSD1 activity leads to Increase, Hepatic intracellular active GC | February 24, 2026 08:32 |
| Increase, Hepatic intracellular active GC leads to Increase, GR activation | February 24, 2026 08:32 |
| Increase, GR activation leads to Increase, Insulin resistance | February 12, 2026 07:38 |
| Increase, Insulin resistance leads to Increase, Adipose lipolysis | February 10, 2026 08:59 |
| Increase, Adipose lipolysis leads to Increased, FA Influx | February 24, 2026 08:33 |
| Increased, FA Influx leads to Increase, Liver steatosis | February 24, 2026 08:33 |
| Increase, Liver steatosis leads to Increase, Hepatocellular lipotoxicity | February 10, 2026 08:59 |
| Increase, Hepatocellular lipotoxicity leads to Increase, ER stress | February 11, 2026 06:16 |
| Increase, ER stress leads to Cell injury/death | February 11, 2026 06:16 |
| Cell injury/death leads to Increase, Kupffer cell activation | November 29, 2016 19:54 |
| Increase, Kupffer cell activation leads to Increase, Steatohepatitis | February 10, 2026 09:00 |
| Increase, Steatohepatitis leads to Activation of TGF-β signaling | February 10, 2026 09:00 |
| Activation of TGF-β signaling leads to Increase, HSC activation | February 10, 2026 09:01 |
| Increase, HSC activation leads to Increase, Collagen accumulation | December 05, 2018 08:51 |
| Increase, Collagen accumulation leads to Increase, Liver fibrosis | December 05, 2018 08:52 |
| Increase, Liver fibrosis leads to Increase, Regenerative nodule formation | February 10, 2026 09:02 |
| Increase, Regenerative nodule formation leads to Increase, Cirrhosis | February 10, 2026 09:02 |
Abstract
This adverse outcome pathway (AOP) describes a mechanistic sequence linking altered glucocorticoid receptor (GR) signaling to the progression of metabolic dysfunction–associated steatotic liver disease (MASLD) through suppression of hepatic de novo lipogenesis (DNL) and subsequent mitochondrial dysfunction. Disruption of GR signaling reduces coordinated lipogenic and lipid-buffering pathways in hepatocytes, impairing the safe esterification and storage of fatty acids. This imbalance promotes hepatocellular lipotoxicity, mitochondrial dysfunction, oxidative stress, and hepatocyte injury, triggering inflammatory and fibrogenic responses. Activation of Kupffer cells, hepatic stellate cells, and TGF-β signaling drives extracellular matrix deposition and fibrotic remodeling, ultimately leading to steatohepatitis (MASH), fibrosis, and cirrhosis. This AOP provides a biologically plausible and complementary framework to support the identification and prioritization of endocrine-disrupting chemicals (EDCs) that interfere with GR-regulated lipid homeostasis and contribute to MASLD progression.
AOP Development Strategy
Context
While increased de novo lipogenesis is commonly associated with hepatic steatosis, emerging evidence indicates that suppression of lipogenic pathways can also exacerbate liver injury by limiting the hepatocyte’s capacity to safely process and store fatty acids. De novo lipogenesis contributes not only to triglyceride synthesis but also to lipid remodeling and buffering processes that protect against lipotoxic species accumulation.
Glucocorticoid receptor (GR) signaling regulates key transcriptional programs involved in hepatic lipogenesis, fatty acid handling, and mitochondrial metabolism. Altered GR signaling—due to dysregulation or chemical interference—can reduce DNL, disrupt lipid homeostasis, and shift hepatocytes toward accumulation of toxic lipid intermediates. This AOP was developed to capture this less intuitive but mechanistically important route linking altered GR signaling to MASLD progression.
Strategy
The AOP was developed through expert-driven pathway conceptualization supported by targeted literature evaluation in the areas of endocrine regulation, hepatic lipid metabolism, and chronic liver disease. Initial scoping identified reduced DNL as a plausible upstream disturbance capable of promoting hepatocellular lipotoxicity independent of increased lipid influx.
Focused literature searches were conducted to identify evidence supporting:
-
GR regulation of hepatic lipogenic gene networks
-
Consequences of suppressed DNL on lipid buffering and toxicity
-
Links between lipotoxicity, mitochondrial dysfunction, and oxidative stress
-
Inflammatory and fibrogenic signaling driving disease progression
Evidence from human clinical studies, animal models, and mechanistic in vitro systems was evaluated for biological plausibility, consistency, and relevance to regulatory toxicology, particularly in the context of chronic, low-dose chemical exposure.
Summary of the AOP
Events:
Molecular Initiating Events (MIE)
Key Events (KE)
Adverse Outcomes (AO)
| Type | Event ID | Title | Short name |
|---|
| MIE | 2410 | Increase, 11β-Hydroxysteroid dehydrogenase type 1 activity | Increase, 11β-HSD1 activity |
| KE | 2412 | Increase, Hepatic intracellular active glucocorticoids | Increase, Hepatic intracellular active GC |
| KE | 122 | Increase, Glucocorticoid receptor activation | Increase, GR activation |
| KE | 2119 | Increase, Insulin resistance | Increase, Insulin resistance |
| KE | 2408 | Increase, Adipose lipolysis | Increase, Adipose lipolysis |
| KE | 465 | Increased, Fatty acid influx | Increased, FA Influx |
| KE | 2405 | Increase, Hepatocellular lipotoxicity | Increase, Hepatocellular lipotoxicity |
| KE | 1815 | Increase, Endoplasmic reticulum stress | Increase, ER stress |
| KE | 55 | Increase, Cell injury/death | Cell injury/death |
| KE | 134 | Increase, Kupffer cell activation | Increase, Kupffer cell activation |
| KE | 1271 | Increase, Transforming growth factor-beta signaling | Activation of TGF-β signaling |
| KE | 265 | Increase, Hepatic stellate cell activation | Increase, HSC activation |
| KE | 68 | Increase, Collagen accumulation | Increase, Collagen accumulation |
| KE | 2406 | Increase, Regenerative nodule formation | Increase, Regenerative nodule formation |
| AO | 459 | Increase, Liver steatosis | Increase, Liver steatosis |
| AO | 1489 | Increase, Steatohepatitis | Increase, Steatohepatitis |
| AO | 344 | Increase, Liver fibrosis | Increase, Liver fibrosis |
| AO | 2407 | Increase, Cirrhosis | Increase, Cirrhosis |
Relationships Between Two Key Events (Including MIEs and AOs)
| Title | Adjacency | Evidence | Quantitative Understanding |
|---|
Network View
Prototypical Stressors
Life Stage Applicability
Taxonomic Applicability
Sex Applicability
Overall Assessment of the AOP
This AOP is biologically plausible and supported by moderate to strong empirical evidence demonstrating that impaired lipogenic capacity can exacerbate lipotoxic stress and liver injury. The sequence of events reflects conserved cellular stress responses and fibrogenic mechanisms observed across mammalian species.
The AOP is particularly relevant for hazard identification and prioritization of chemicals that interfere with GR signaling and hepatic lipid homeostasis but may not induce classical steatogenic responses. While quantitative understanding across the full pathway is limited, the overall weight of evidence supports its inclusion within an AOP network describing GR-mediated MASLD progression.
Domain of Applicability
-
Taxa: Mammals (humans and laboratory rodents)
-
Life stage: Primarily adolescents and adults
-
Sex: Applicable to both sexes, with potential sex-dependent modulation of lipogenic responses
-
Biological context: Chronic endocrine perturbation, metabolic stress, or impaired lipid handling
This AOP is not intended to describe acute liver toxicity and is most applicable to chronic exposure scenarios.
Essentiality of the Key Events
Evidence supporting the essentiality of the key events includes:
-
Altered GR signaling: Experimental manipulation of GR activity alters hepatic lipogenic programs and lipid handling capacity.
-
Reduced de novo lipogenesis: Suppression of DNL limits safe fatty acid esterification, increasing susceptibility to lipotoxic injury.
-
Hepatocellular lipotoxicity and mitochondrial dysfunction: Restoration of lipid buffering or mitochondrial function attenuates oxidative stress and cell injury.
-
Inflammatory and fibrogenic activation: Inhibition of Kupffer cell activation, hepatic stellate cell activation, or TGF-β signaling reduces fibrosis progression in experimental models.
These findings support the essential role of each KE in driving downstream MASLD outcomes.
Evidence Assessment
Across the KERs in this AOP:
-
Biological plausibility is strong, based on established roles of GR signaling and lipogenic pathways in hepatocyte homeostasis.
-
Empirical support is moderate, with increasing evidence linking reduced DNL to lipotoxicity and mitochondrial stress.
-
Quantitative understanding is limited to individual relationships, with few studies integrating effects across multiple downstream events.
Overall, the weight of evidence supports confidence in the pathway for regulatory-relevant applications.
Known Modulating Factors
| Modulating Factor (MF) | Influence or Outcome | KER(s) involved |
|---|---|---|
| Dietary fatty acid composition | Modulates reliance on DNL for lipid buffering | DNL ↓ → lipotoxicity |
| Insulin signaling status | Influences lipogenic gene expression | GR signaling → DNL |
| Mitochondrial capacity | Alters susceptibility to oxidative stress | Lipotoxicity → mitochondrial dysfunction |
| Inflammatory milieu | Amplifies hepatocyte injury and fibrosis | Oxidative stress → fibrosis |
Quantitative Understanding
Quantitative data exist for individual steps linking reduced DNL to altered lipid profiles and hepatocellular stress. However, quantitative integration across mitochondrial, inflammatory, and fibrotic events is currently limited. This AOP is therefore best applied qualitatively or semi-quantitatively.
Considerations for Potential Applications of the AOP (optional)
This AOP may support:
-
Identification of GR-modulating chemicals that disrupt hepatic lipid buffering
-
Complementary assessment of non-steatogenic mechanisms contributing to MASLD
-
Development of integrated testing strategies incorporating lipid handling and mitochondrial endpoints
-
AOP network construction capturing multiple GR-mediated routes to MASLD progression