AOP ID and Title:
Graphical Representation
Status
| Author status | OECD status | OECD project | SAAOP status |
|---|---|---|---|
| Under Development: Contributions and Comments Welcome | Under Development | 1.105 | Included in OECD Work Plan |
Abstract
Breast cancer is the deadliest cancer in women with a poor prognosis in case of metastatic breast cancer. The role of the environments in the formation of metastasis has been suggested. We hypothesized that activation of the AhR (MIE), a xenobiotic receptor, could lead to breast cancer metastasis (AO), through different KEs, constituting a new AOP.
An artificial intelligence tool (AOP-helpfinder), which screens the available literature, was used to collect all existing scientific abstracts to build a novel AOP, using a list of key words. Four hundred and seven abstracts were found containing at least a word from our MIE list and either one word from our AO or KE list. A manual curation retained 113 pertinent articles, which were also screened using PubTator. From these analyses, an AOP was created linking the activation of the AhR to breast cancer related death through decreased apoptosis, inflammation, endothelial cell migration, and increased mortality. These KEs promote an increased tumor growth, angiogenesis and invasion which leads to breast cancer metastasis.
The evidence of the proposed AOP was weighted using the tailored Bradford Hill criteria and the AOP developers’ handbook (https://aopwiki.org/handbooks/). The confidence in our AOP and the biological plausibility was considered strong. Indeed, in vitro and in vivo findings on multiple types of breast cancers (with or without oesrtogen receptors, for instanace) supported our proposed AOP. An in vitro validation must be carried out, but our review proposes a strong relationship between AhR activation and breast cancer metastasis with an innovative use of an artificial intelligence literature search.
This work was published in Envionnmental International: https://doi.org/10.1016/j.envint.2022.107323
Background
Breast cancer is a frequent disease, responsible of 2 262 419 new cases and 684 996 deaths in 2020 in the world, making it the deadliest female cancer (Bray et al., 2018). In 70% of cases, the disease is localized, and the prognosis is favorable with a 5-year survival of 99%. However, once the disease spreads (lymph nodes, metastasis), survival is severely altered with a 5-year survival rate of 26% in case of metastasis (Henley et al., 2020). It is therefore of paramount importance to understand the mechanisms of metastasis in breast cancer.
Amongst risk factors clearly established, including obesity, genetic mutations and hormonal exposure, the importance of the role of the environment is currently emerging (Koual et al., 2020 Nov 17). In an epidemiologic study, we found a positive association between the concentrations of 2.3.7.8-TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxine) in the adipose tissue surrounding the tumors, and breast cancer metastasis in overweight and obese patients (Koual et al., 2019). Moreover, we have shown that, using both in vivo and in vitro models, TCDD exposure could promote an aggressive phenotype to breast cancer cells, thus favoring the formation of metastatic cells (Koual et al., 2021). TCDD is a potent ligand of the aryl hydrocarbon receptor (AhR), a transcriptional factor involved notably in the metabolism of xenobiotics (Larigot et al., 2022). Hence, the impact of the environment on breast cancer aggressiveness could be mediated by the activation of the AhR.
Interest is growing on the role of the AhR in breast cancer. First, the AhR is often overexpressed in different breast cancer cell lines (Zudaire et al., 2008, Kim et al., 2000 Nov 16, Li et al., 2014). Interestingly, the level of expression can be correlated to the stage or the molecular sub-type of the disease (Zudaire et al., 2008, Zhao et al., 2013). Second, the AhR pathway has been associated with different pro-metastatic features in breast cancer, such as resistance to apoptosis, invasiveness, modified cell cycle, migration and proliferation (Zudaire et al., 2008, Goode et al., 2013 Dec 15, Kanno et al., 2006). Triple negative cell lines, breast cancer cell lines with the worse prognosis (not over-expressing Her2 receptor or hormonal receptors), over-expressing the AhR seem to develop stem-like characteristics, favoring epithelial-mesenchymal transition (EMT) and thus metastasis (Stanford et al., 2016). Thirdly, the AhR could be involved in the resistance of breast cancer to treatments (Goode et al., 2013 Dec 15, Goode et al., 2014): after AhR knockout, Goode et al. found enhanced sensitivity of paclitaxel (a drug targeting cancer cells) in triple negative breast cancer, a cancer particularly difficult to treat (Goode et al., 2014). Breast cancer patients expressing estrogen receptors (ER-positive) in their cancer cells, can benefit from an efficient endocrine therapy, which greatly improves their survival. Activation of the AhR can lead to the loss of expression of the ER alpha and therefore to the loss of a potential therapeutic target (Safe et al., 2000 Jul).
The mechanisms linking the activation of the AhR to breast cancer aggressiveness are still unclear. Based on the AOP-wiki database (https://aopwiki.org/, last accessed March 2022), the central repository for AOPs, the AhR has already been proposed in several AOPs, but never in one characterized by the AO breast cancer metastasis. Likewise, an AOP linking an MIE to breast cancer aggressiveness has never been proposed. From our expertise and available knowledge, we hypothesize that the activation of the AhR could be a MIE leading to breast cancer metastasis (AO) through different KEs and KERs.
Summary of the AOP
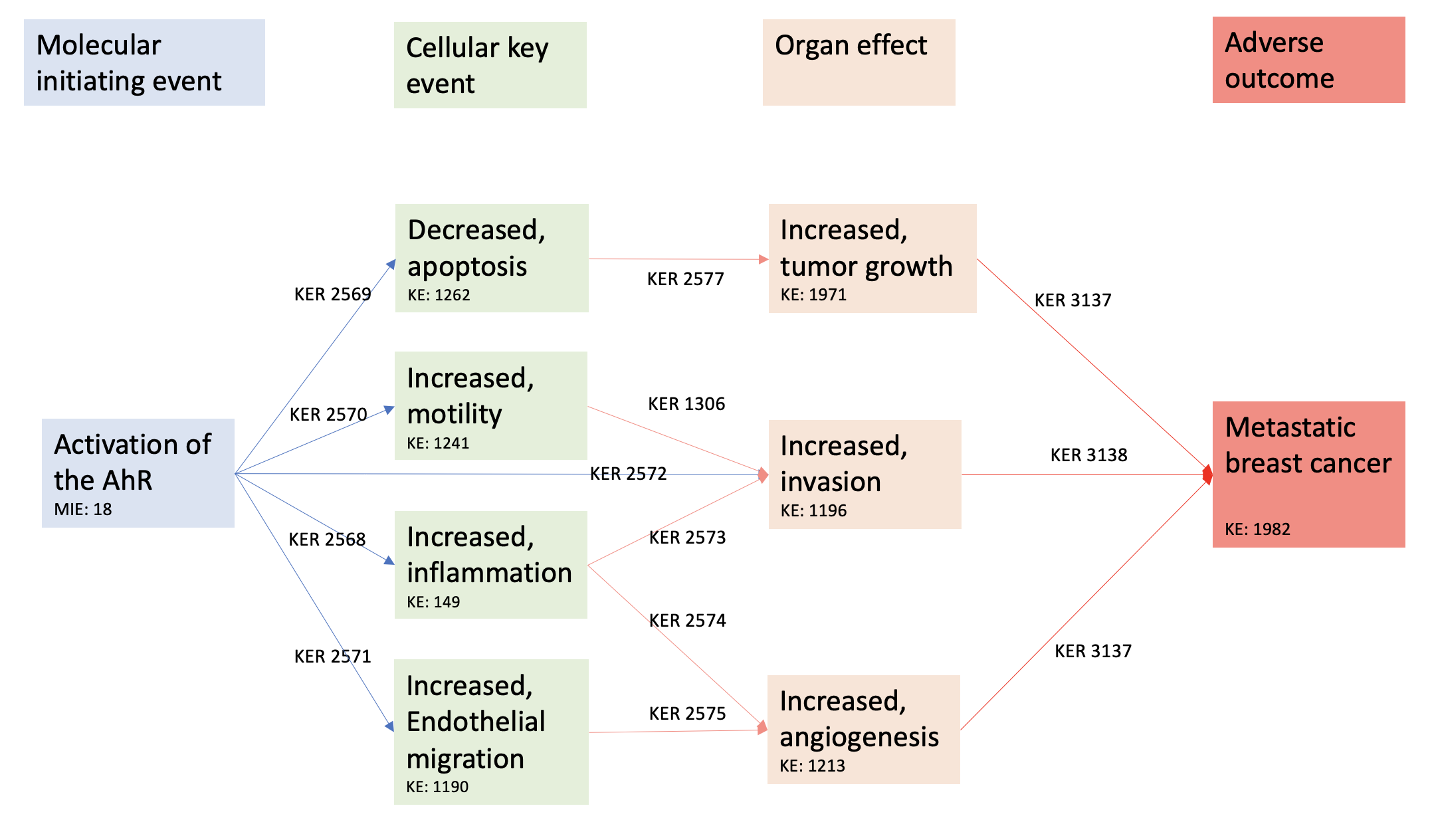
Events
Molecular Initiating Events (MIE), Key Events (KE), Adverse Outcomes (AO)
| Sequence | Type | Event ID | Title | Short name |
|---|---|---|---|---|
| MIE | 18 | Activation, AhR | Activation, AhR | |
| KE | 149 | Increase, Inflammation | Increase, Inflammation | |
| KE | 1262 | Apoptosis | Apoptosis | |
| KE | 1241 | Increased, Motility | Increased, Motility | |
| KE | 1190 | Increased, Migration (Endothelial Cells) | Increased, Migration (Endothelial Cells) | |
| KE | 1196 | Increased, Invasion | Increased, Invasion | |
| KE | 1376 | Increase, angiogenesis | Increase, angiogenesis | |
| KE | 1971 | Increased, tumor growth | tumor growth | |
| AO | 1982 | metastatic breast cancer | Metastasis, Breast Cancer |
Key Event Relationships
| Upstream Event | Relationship Type | Downstream Event | Evidence | Quantitative Understanding |
|---|---|---|---|---|
| Activation, AhR | adjacent | Increase, Inflammation | High | |
| Activation, AhR | adjacent | Apoptosis | High | |
| Activation, AhR | adjacent | Increased, Motility | High | |
| Activation, AhR | adjacent | Increased, Migration (Endothelial Cells) | Moderate | |
| Activation, AhR | adjacent | Increased, Invasion | High | |
| Increase, Inflammation | adjacent | Increased, Invasion | High | |
| Increase, Inflammation | adjacent | Increase, angiogenesis | High | |
| Increased, Motility | adjacent | Increased, Invasion | High | |
| Increased, Migration (Endothelial Cells) | adjacent | Increase, angiogenesis | High | |
| Apoptosis | adjacent | Increased, tumor growth | High | |
| Increase, angiogenesis | adjacent | metastatic breast cancer | High | High |
| Increased, Invasion | adjacent | metastatic breast cancer | High | High |
| Increased, tumor growth | adjacent | metastatic breast cancer | High | High |
Stressors
| Name | Evidence |
|---|---|
| 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) |
Overall Assessment of the AOP
The biological plausibility of KERs is defined by the OECD as the « understanding of the fundamental biological processes involved and whether they are consistent with the causal relationship being proposed in the AOP ». The biological plausibility is strong due to the presence of overwhelming evidence present in different studies. A minor setback would be the difficulty to dismiss alternative mechanisms caused by the ligands used for AhR activation. This is detailed in the discussion.
The essentiality of KEs refers to « experimental data for whether or not downstream KEs or the AO are prevented or modified if an upstream event is blocked ». The essentiality of KEs is strong: most works use suppression or inhibition of the AhR (knock out, antagonists and/or silencing) with results coherent with our findings.
Finally, the empirical support of KERs, is often « based on toxicological data derived by one or more reference chemicals where dose–response and temporal concordance for the KE pair can be assessed ». The overall assessment of the empirical support of our KERs is also strong. There is evidence in human cell lines and mice showing a dose–response and temporal concordance for severity of our KE and the presence of metastasis.
We propose a simple and robust AOP associating activation of the AhR and breast cancer related death through migration, invasion, inflammation, and neo-angiogenesis.
One of the main limitations of our AOP is the existence of these diverse ligands and pathways, complexifying the definition of ‘AhR activation’ (6,54). Using PubTator, we found that TCDD was by far the most used chemical followed by I3C, alpha-naphthoflavone, polycyclic aromatic hydrocarbons and hexachlorobenzene, all ligands of the AhR. These ligands can activate different pathways after AhR binding and we therefore assumed that these compounds were AhR agonists. It can be difficult to dismiss alternative mechanisms caused by the ligands used for AhR activation. However, the AhR is the only characterized target of TCDD for example, and studies which use several ligands including TCDD, display similar results using the other modulators. Moreover, the concordance of studies using various ligands and the coherence with the AhR inhibition are in favor of the robustness of the proposed AOP. Indeed, to obtain the most accurate AOP possible, the KEs selected had to be present, no matter the ligand used by the study.
Another minor setback of using the AhR, is that the dose response concordance is a non-monotonous curve for several ligands (122,123). Therefore, the tailored Bradford-Hill criteria could sometimes not be fulfilled.
Moreover, the originality of our work lies in the use of artificial intelligence too such as AOP-helpfinder, which enables a thoroughly search of existing knowledge in the PubMed database and PubTator (19–21). Therefore, our literature review was complete and evidence in favor of our proposed AOP was overwhelming. We plan to validate our proposed AOP in a quantitative in vitro work using Integrated Approaches to Testing and Assessment (IATA).
Domain of Applicability
Life Stage Applicability| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Female | High |
| Male | Low |
The biological applicability domain of the putative AOP concerned mainly females of menstrual of post-menopausal age. Indeed, existing cell lines were derived from women of menstrual of post-menopausal age and in vivo, studies were performed on mice of reproductive age. Only one study used the zebra fish larvae (Narasimhan et al., 2018 May 7). However, it could be extrapolated to men. Indeed, breast cancers in men present similar tumor characteristics and no work has found diverging functions of the AhR between men and women. Moreover, no difference in AhR expression has been characterized between men and women. Furthermore, our AOP concerns ER-positive and triple negative cells lines.
Studies were carried out in humans, mice, and zebrafish (xenotransplant studies, no mammary gland) (i.e. PubTator results) and it can be hypothesized that this AOP is conserved across mammals. Indeed, the AhR is a very conserved and ancient protein (Hahn, 2002 Sep 20). However, since the sensitivity to adverse events are variable among taxa, we can only postulate this AOP in human and mice (Korkalainen et al., 2001 Aug 3, Cohen-Barnhouse et al., 2011 Jan, Doering et al., 2013 Mar).
The AhR is a fascinating yet complex receptor since its activation is ligand and cell dependent. To avoid more bias, we decided to limit our AOP to breast cancer. First, this cancer is the most frequent female malignancy, which makes it a major public health concern. Second, this illness is hormonal-dependent and therefore the impact of the environment, through the AhR, can be strongly suggested. However, we have reasons to believe this AOP could be extrapolated to other cancers which share common regulatory pathways (Larigot et al., 2022). The AhR is overexpressed not only in breast cancer but also in lung, liver, stomach, head & neck, cervix, and ovarian cancer (Stanford et al., 2016, DiNatale et al., 2010 Aug 6, Liu et al., 2013 Aug, Stanford et al., 2016 Aug). Moreover, in these cancers, the level of expression is correlated to the stage of the disease (Zudaire et al., 2008, Koliopanos et al., 2002 Sep 5, Chang et al., 2007 Jan 1). Additionally, Moenniks et al. found that mice with constitutively active AhR had more liver tumors than wild type mice (55% versus 6%) (Moennikes et al., 2004 Jul 15). In vitro evidence suggests that the AhR activation could promote a more aggressive phenotype to renal, lung, head and neck, and urothelial cancer through an increase in invasion, migration, and resistance to apoptosis which constitute representative key events of our AOP (Zudaire et al., 2008, Stanford et al., 2016 Aug, Ishida et al., 2015 Jul 15, Ishida et al., 2010 Feb, Diry et al., 2006 Sep 7, John et al., 2014 Oct). Besides, an AOP associating AhR activation and lung cancer initiation is currently under development (AOP, 2021) (https://aopwiki.org/aops/417, accessed May 2022).
Likewise, our AOP covers only breast cancer progression and not initiation. The mechanisms of breast cancer initiation are different from the metastatic pathway, but the AhR could also be involved in breast cancer initiation. In vitro, it was noted that human mammary benign cells with a high level of AhR had an increase in cell proliferation, and migration, and potentially display EMT-like features (Brooks and Eltom, 2011 Jun). In vivo, mice fed with 7,12-dimethylbenz[a]anthracene (DMBA, an AhR activator and a potent mutagen) had an increased risk of mammary tumors, with higher AhR expression (Currier et al., 2005). Strangely in regard of the deadly outcomes associated with aggressive breast tumors, the number of studies focusing on this specific aspect of mammary carcinogenesis is limited and therefore, epidemiological data on the effects of the exposome in breast cancer aggressiveness is scarce. Indeed, occupational exposure is difficult to quantify, and patients are usually exposed to a mixture of pollutants and not a single pollutant in a chronic way. A memory bias cannot be excluded since the half-life of TCDD, for instance, is 7–11 years (Pirkle et al., 1989). Industrial accidents, such as the Seveso incident, studied the increase in breast cancer incidence but did not record breast cancer aggressiveness since it is more complex to quantify. At an early stage, breast cancer has a favorable prognosis whereas the therapeutic challenge lies in the treatment of breast cancer metastases. Therefore, even though epidemiologic and cell evidence suggests that exposure to pollutants and Ahr activation could promote breast cancer initiation, we chose to study breast cancer progression, the most complex situation (Pesatori et al., 2009 Sep, Warner et al., 2002 Jul).
Essentiality of the Key Events
| KEY EVENT | LEVEL OF ESSENTIALITY | EVIDENCE | ||
| KE 1262 : decreased apoptosis | Strong |
A decrease in apoptosis is an essentiel element in promoting tumor growth (Hannahan , Fulda). Indeed, in case of a decrease in cell death, the tumor will continue to grow. First, A decrase in apoptosis causes uncontrolled Cell Proliferation. Healthy tissues maintain homeostasis through a balance between cell proliferation and apoptosis. When apoptosis is compromised, cells that should undergo programmed cell death survive and continue dividing. This leads to an unchecked increase in cell number, forming the initial tumor mass. [Hanahan & Weinberg, 2011]. Second, cancer cells sustain proliferative signaling. Many cancers harbor mutations that activate pro-proliferative signaling pathways like Ras or PI3K/Akt. These pathways normally promote cell growth and division. However, mutational dysregulation allows them to continue signaling proliferation even when apoptosis should occur or growth signals are absent. Additionally, reduced apoptosis prevents the activation of pro-apoptotic pathways that normally act as brakes on cell division. [Luo & Heng, 2003] Also, healthy cells respond to cues like density-dependent inhibition and nutrient limitations by activating apoptosis. When apoptosis is compromised, cells can evade these growth-inhibitory signals and continue dividing even when resources are limited or cell density is high. This allows the tumor to expand beyond its boundaries and invade surrounding tissues. [Fulda & Debatin, 2007]. A decrease in apoptosis is therefore essential to maintain tumor growth. However, cell proliferation is also an essentiel element in promoting tumor growth. Yet, due to the presence of diverging evidence on the activation of the AhR and cell proliferation, we chose not to include these in our AOP. Indeed, on one hand, activation of the AhR through ligands such as NK150460, ANI-7, emodine or derivates of revesterol decrease cell proliferation in ER-positive and ER-negative breast cancer cell lines. TCDD has been found to promote cell cycle arrest through phosphorylation of the retinoblastoma protein which binds to E2F. In ER-positive cell lines, beta-naphthoflavone mediated cell cycle arrest through an upregulation of P21. On the other hand, AhR activation could promote cell proliferation. Pearce et al. found that MCDF (6-methyl-1,3,8-trichlorodibenzofuran), an AhR agonist could stimulate cell proliferation with a dose-response concordance. Likewise, I3C, HCB, CPF and licorice could also promote cell proliferation. However, it seems that this cell proliferation is ER-dependent. Indeed, these ligands induced cell proliferation only in ER-positive cells lines with an effect dependent on the level of estrogen present in the medium. Whether this increase in ER-dependent cell proliferation can be independent of the AhR remains unclear. This increase in proliferation could also be mediated by the association of the RelA subunit of NF-kappaB with the AhR resulting in the activation of c-myc gene transcription in breast cancer cells. This would explain why Rodriguez et al. found that proliferation was modulated by the CYP1A1, independently of an exogenous ligand activation of the AhR. These complex effects, highly dependent on the context (cell types, medium content, type of ligand…) were therefore not included in our AOP despite the strong evidence. |
||
| KE 1971 : tumor growth | STRONG | An increase in tumor size is associated with breast cancer metastasis and is essential to the progression of the illness (Hanahan and Weinberg, 2011 Mar 4). Indeed, clinical evidence suggests that tumor size is directly correlated to the presence of metastasis (Liu Y, He M, Zuo WJ, Hao S, Wang ZH, Shao ZM. Tumor Size Still Impacts Prognosis in Breast Cancer With Extensive Nodal Involvement. Front Oncol and Narod SA. Tumour size predicts long-term survival among women with lymph node-positive breast cancer. Curr Oncol.) Likewise, studies have shown that larger tumor size in colorectal cancer is associated with increased risk of metastasis and poorer overall survival. [Benson et al., 2008] | ||
|
KE 1241 Increased cell motility |
MODERATE | The relation between cell migration and organ invasion is essntial. Organ invasion can be promonted by cell migration, motility and inflammation. Therefore the essentilality of cell motility was classified as moderate since other factors can promote organ invasion. For instance, melanoma cells are known for their high migratory potential, allowing them to invade the surrounding dermis and potentially metastasize to distant organs like the brain and lungs. [Clark et al., 2009] Likewise, breast cancer cells can migrate through the basement membrane and invade surrounding breast tissue, potentially reaching lymph nodes or blood vessels for further dissemination. [Friedl & Weigelin, 2008] | ||
|
KE 1196: organ invasion |
STRONG | Organ invasion is an essential step in promoting breast cancer agressivness and metastasis. Without invasion of the basal membrane, the cancer remains located in an in situ state and does not induce metastasis. Pancreatic cancer cells are notorious for their invasive nature. They can invade surrounding tissues like the pancreas, blood vessels, and nerves, increasing the risk of metastasis to the liver, lungs, and bones. [Olive et al., 2009] Colorectal cancer cells can invade the bowel wall and potentially reach surrounding blood vessels, allowing them to travel to the liver, lungs, and other distant sites. [Fearon & Vogelstein, 1990] | ||
| KE 149 Increased inflammation | MODERATE |
Organ invasion can be promonted by cell migration, motility and inflammation. Therefore the essentilality of cell motility was classified as moderate since other factors can promote organ invasion. In angiogenesis, however, increased inflammation is a key factor. Indeed, inflammation, through the secretion of growth factor promotes the creation of blood vessels (VEGF, IL6, COX). |
||
| KE 1190 Increased endothelial migration | STRONG | Endothelial cell migration is an essential key event in promoting angiogenesis. Extensive data exists on the essentialitty of this step (Franziska van Zijl, Georg Krupitza, Wolfgang Mikulits, Initial steps of metastasis: Cell invasion and endothelial transmigration, Mutation Research/Reviews in Mutation Research, Volume 728, Issues 1–2, 2011, Pages 23-34, ISSN 1383-5742, https://doi.org/10.1016/j.mrrev.2011.05.002.) | ||
| KE 1213: angiogenesis | STRONG | Without the creation of new vessels in order to receive nutrients and energy, the cancer cell cannot survive and create metastatis. It is an essential key event and considered as one of the hallmarks of cancer (Hanahan and Weinberg, 2011 Mar 4). |
Weight of Evidence Summary
KER 2569 Activation of the AhR leads to decreased apoptosis
Several studies have found that the activation of the AhR by stressors such as TCDD, can promote a decrease in apoptosis (KER2569), which is a deleterious event with regards to cancer (Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015). Additionally, an increase in cell death was found when blocking the AhR pathway using AhR silencing (RNA interference or knock-out), knockout cell lines or antagonists (CH223191 or alpha-naphthoflavone) (Goode et al., 2013 Dec 15, Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015, Regan Anderson et al., 2018). The most frequently used assay to evaluate apoptosis was cytometry with the use of Annexin V: this was performed with ER-positive cells lines (MCF-7, T-47D), triple negative cell lines (MDA-MB-231, HS 578), cells over-expressing the Her2 (SK-BR-3) and cells lines derived from cancer samples from patients (Goode et al., 2013 Dec 15, Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015, Regan Anderson et al., 2018, Fujisawa et al., 2011).
The concordance of the evidence was classified as “moderate” since the aim of most studies was to evaluate the capacity to survive in an apoptosis-promoting environment (i.e., chemotherapeutic drugs). Indeed, they assessed the resistance to chemotherapy agents such as doxorubicin and paclitaxel and found that the concomitant inactivation of the AhR pathway could decrease the resistance to these chemotherapy agents through an increase in cell death when compared to cells with a functional (or expressed at sufficient levels) AhR (Goode et al., 2013 Dec 15, Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015, Regan Anderson et al., 2018, Fujisawa et al., 2011). Since the environment was modified by the presence of chemotherapy, the hypothesis of an alternative pathway cannot be completely discarded. It must be noticed that the exact biological mechanisms linking the activation of the AhR to the decrease in apoptosis remains unclear. Indeed, Anderson et al. suggested that the AhR interacts with the glucocorticoid receptor (GR) and the hypoxia inducible factor-2α (HIF-2α) (Regan Anderson et al., 2018). The presence of the GR is associated with a poor prognosis, notably in triple negative breast cancer (Pan et al., 2011, Moran et al., 2000 Feb 15). Indeed, this receptor is involved in survival and resistance to chemotherapy through up-regulation of c-myc, Bcl2 and Kruppel-like factor 5 (Pan et al., 2011, Wu et al., 2004, Li et al., 2017). Both GR and HIF 2α could be up regulated by the AhR. They then activate Brk (also known as PTK6), a ligand of EGFR (epidermal growth factor receptor), involved in the inhibition of apoptosis (Regan Anderson et al., 2018, Li et al., 2012). Another possible mechanism suggested by Bekki et al. is that the decrease in apoptosis was caused by the induction of cyclooxygenase 2 (COX-2) and the NF-κB subunit RelB (Bekki et al., 2015). They both prevent apoptosis through induction of Bcl2, an anti-apoptotic factor (Tsujii and DuBois, 1995, Vogel et al., 2007, Thomas et al., 2020, Baud and Jacque, 2008 Dec, Demicco et al., 2005 Nov, Wang et al., 2007 Apr, Liu et al., 2001 May 25).
KER 2577: Decreased apoptosis promotes tumor growth
For KER 2577, in vivo, Goode et al. showed that the knockout of the AhR in mice reduced tumor growth through an increase of cell apoptosis (Goode et al., 2013 Dec 15).
The relationship between decreased apoptosis and increase in tumor growth (KER 2577) is not detailed here due to extensive evidence in the scientific literature (Hanahan and Weinberg, 2011 Mar 4).
KER 2570: Activation of the AhR leads to an increased cell motility
The activation of the AhR can modulate cell motility in different types of breast cancers such as: ER-positive cells lines (MCF-7, T-47D, ZR-75–1), triple negative (MDA-MB-231, MDA-MB-435, HS-578-T, SUM149), and cells overexpressing the Her2 (SK-BR-3) (Goode et al., 2013 Dec 15, Regan Anderson et al., 2018, Parks et al., 2014 Nov, Pontillo et al., 2011 Apr, Qin et al., 2011 Oct 20, Nguyen et al., 2016 Nov 15, Novikov et al., 2016 Nov, Miret et al., 2016 Jul, Shan et al., 2020 Nov, Dwyer et al., 2021 Feb, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb). Activation of the AhR with TCDD, butyl-benzyl phthalate, di-n-butyl phthalate, hexachlorobenzene, and benzo[a]pyrene can promote cell migration in different assays (Parks et al., 2014 Nov, Pontillo et al., 2011 Apr, Qin et al., 2011 Oct 20, Novikov et al., 2016 Nov, Miret et al., 2016 Jul, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb). On the other hand, the use of AhR antagonists, AhR silencing or AhR knockout reversed this effect (Goode et al., 2013 Dec 15, Regan Anderson et al., 2018, Parks et al., 2014 Nov, Pontillo et al., 2011 Apr, Qin et al., 2011 Oct 20, Novikov et al., 2016 Nov, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb). The most frequently used assays for evaluating cell migration were the scratch wound assay and the transwell chamber assay. Only three works evaluated the dose–response concordance of AhR activation with stressors and cell migration (Pontillo et al., 2011 Apr, Miret et al., 2016 Jul, Shan et al., 2020 Nov). The evidence was therefore classified as “moderate”.
KER 2572: Activation of the AhR leads to an increased invasion
Due to the extensive robust and concordant literature of the link between activation of the AhR-increased cell motility-increased invasion-breast cancer progression, the confidence in these key events was rated as high. However, due to the use of ligands to activate the AhR, it cannot be completely ruled out that alternative pathways (independent of the AhR) can also contribute to these features. For instance, 2 main pathways seem to explain this increase in migration and invasion: the c-Src/HER1/STAT5b, and ERK1/2 pathways. Yet, these pathways seem only to explain the relation between the AhR activation and cell migration / invasion, when the ligand used is hexachlorobenzene, an organochlorinated pesticide (Pontillo et al., 2011 Apr, Miret et al., 2016 Jul, Pontillo et al., 2013 May 1). Even though alternative mechanisms may present themselves, all studies blocked the AhR pathway and found a decrease in cell migration/invasion. The evidence for alternative mechanisms was therefore classified as “moderate” and the biological plausibility of KER was also classified as “moderate”.
KER 1306: Increased cell motility promotes organ invasion
The relation between cell migration and organ invasion has already been shown (KER-1306, https://aopwiki.org/relationships/1306). Since the 2 are closely linked, most articles studied both cell migration (chemo-tactic) and the capacity to invade the extra-cellular matrix. Cell invasion is indeed defined as the capacity of a cell to migrate and degrade/invade the extracellular matrix. In vitro, this process was evaluated mostly using transwell chamber with Matrigel® and the presence of matrix metalloproteinases (MMP). This effect was found in ER-positive cells, triple negative cell lines and cells overexpressing the Her2.
KER 2572: Activation of the AhR leads to an increased invasion
The activation of the AhR through the use of different ligands (benzophenone, butyl benzyl phthalate, di-n-butyl phthalate, hexachlorobenzene, chlorpyrifos, TCDD) or the blockage of the AhR (silencing, KO or antagonism) increased or decreased cell invasion, respectively (Parks et al., 2014 Nov, Qin et al., 2011 Oct 20, Nguyen et al., 2016 Nov 15, Miret et al., 2016 Jul, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb, Pontillo et al., 2013 May 1, Miller et al., 2005, Belguise et al., 2007 Dec 15, Yamashita et al., 2018 May 1, Miret et al., 2020 May). The dose–response concordance for cell invasion was demonstrated using increasing doses of hexachlorobenzene, benzo[a]pyrene, chlorpyrifos and TCDD (Miret et al., 2016 Jul, Shan et al., 2020 Nov, Pontillo et al., 2013 May 1, Miller et al., 2005, Miret et al., 2020 May). To further explore cell invasion, Nguyen et al. created a model of a lymphatic barrier using a three-dimensional lymph endothelial cell as a monolayer co-cultured with spheroids of MDA-MB231 cells (Nguyen et al., 2016 Nov 15). They found that silencing or antagonizing the AhR (DIM) or activating the AhR (FICZ) respectively decreased or increased invasion of the lymphatic barrier.
On an organ level, in vivo, an increase in metastasis has been found in mice and zebrafish after the activation of the AhR with different ligands (butyl benzyl phthalate, di-n-butyl phthalate, hexachlorobenzene, TCDD) (Goode et al., 2014, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb, Pontillo et al., 2013 May 1). In the zebrafish model, Narasimham et al. treated the animals either with triple negative MDA-MB-231 cells only (untreated) or with MDA-MB-231 cells treated with an AhR inhibitor (CB7993113 or CH22319) (Narasimhan et al., 2018 May 7). Untreated fish had significantly more metastasis (OR = 9, IC95%=3–35). Similar results were found using mice models (Goode et al., 2014, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb, Pontillo et al., 2013 May 1).
KER 2568: Activation of the AhR leads to an increased inflammation
In triple negative breast cell lines (MDA-MB436, MDA-MB-231) and ER-positive cell lines, it has been shown that the activation of the AhR can lead to an increase in inflammation. (Bekki et al., 2015, Miller et al., 2005, Yamashita et al., 2018 May 1, Degner et al., 2009 Jan, Vogel et al., 2011 Aug 1, Kolasa et al., 2013 Apr 25, Vacher et al., 2018, Malik et al., 2019 Oct). The stressors mainly used to activate the AhR were TCDD followed by benzo[a]pyrene and 2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine (PhiP). After AhR inhibition (KO or antagonists), a decrease in inflammation biomarkers was found (Miller et al., 2005, Yamashita et al., 2018 May 1, Degner et al., 2009 Jan, Vogel et al., 2011 Aug 1, Kolasa et al., 2013 Apr 25). Assays evaluating cell inflammation were quantitative dosages of IL-6, IL-8 and Cox2 activity/expression. Cox-2 and IL-8 were amongst the top “gene concepts” retrieved by the PubTator Central tool, likewise, “inflammation” was frequently found as a disease concept. The most consensual pathway linking the AhR activation to cell inflammation was the NF-kB pathway (Vogel et al., 2011 Aug 1, Kolasa et al., 2013 Apr 25). Only half of the studies found a dose–response relationship (Miller et al., 2005, Kolasa et al., 2013 Apr 25, Malik et al., 2019 Oct). No studies were carried out in vivo for breast cancer and therefore the concordance and evidence were classified as “moderate”.
AOP 21 also found the association between AhR activation and inflammation via COX 2 (Aryl hydrocarbon receptor activation leading to early life stage mortality, via increased COX-2) with a weight of evidence classified as “high”. Indeed, the AhR/ARNT heterodimer links to the dioxin responsive elements which in turn up-regulates COX-2 (66,67].
KER 2573: Inflammation promotes organ invasion
In the specific setting of AhR activation, only 2 studies showed the continuum between AhR activation – increased inflammation – increased invasion (Miller et al., 2005, Yamashita et al., 2018 May 1). However, in general, there is extensive knowledge on the relationship between cell inflammation and organ invasion. First, COX-2 is expressed at higher levels in triple negative invasive breast cancers than in less aggressive ER-positive cancers (Gilhooly and Rose, 1999 Aug, Liu and Rose, 1996 Nov 15). COX-2 catalyzes the conversion of arachidonic acid into prostaglandin H2, a pro-inflammatory factor, and is therefore considered as a prognosis factor in breast cancer (Ristimäki et al., 2002 Feb 1, Parrett et al., 1997 Mar). Transfection with COX-2 triple negative MDA-MB-435 cells increased cell migration 2-fold compared to control cells in a transwell-Matrigel® assay. Antagonism of COX-2 through an inhibitor (NS-398) reversed this action in a dose-dependent way (Singh et al., 2005 May). Second, in vivo, the use of anti-inflammatory treatments such as celecoxib (COX-2 inhibitor) can reduce tumor growth and spread (Harris et al., 2000 Apr 15). Finally, epidemiologic evidence suggests that inflammatory breast cancers have the worse prognosis. Indeed, the median overall survival of patients with inflammatory breast cancer compared with those with non-inflammatory breast cancer tumors is 4.75 years versus 13.40 years for stage III disease and 2.27 years versus 3.40 years for stage IV disease (Schlichting et al., 2012 Aug, Fouad et al., 2017 Apr).
The mechanism of action of COX-2 are consensual. COX-2 promotes cell invasion through upregulation of MMPs (notably 2 and 9) (Takahashi et al., 1999 Oct 22, Sivula et al., 2005 Feb, Larkins et al., 2006 Jul). Moreover, COX-2 could also activate the urokinase plasminogen activator (uPA) which degrades the basal membrane of epithelia (Singh et al., 2005 May, Takahashi et al., 1999 Oct 22, Larkins et al., 2006 Jul, Guyton et al., 2000 Mar).
The relationship between inflammation and invasion is well document therefore the evidence was classified as “strong”.
KER 2574: Inflammation promotes angiogenesis
Likewise, two studies evaluated the specific continuum AhR activation – increased inflammation – increased angiogenesis (Pontillo et al., 2015 Nov 19, Zárate et al., 2020 Aug). As previously mentioned, the AhR activation increases inflammation, notably through an increase in COX 2 (Bekki et al., 2015, Miller et al., 2005, Degner et al., 2009 Jan, Pontillo et al., 2015 Nov 19, Zárate et al., 2020 Aug).
COX-2 can promote angiogenesis through an increase in VEGF (Vascular endothelial growth factor) (Harris et al., 2014 Oct 10, Kirkpatrick et al., 2002). In a pathologic study characterizing 46 breast cancer specimen using immunochemistry, it was found that the density of microvessels was significantly higher in patients with COX-2 expression than in those without expression (p = 0.03) (Costa et al., 2002 Jun). The relationship between COX-2 and angiogenesis has also been shown in gastric and colorectal cancer (Tsujii et al., 1998 May 29, Uefuji et al., 2000 Jan). Indeed, colon carcinoma cells overexpressing COX-2 produce proangiogenic factors (VEGF, bFGF, TBF-β, PDGF, and endothelin-1), and stimulate endothelial migration and the formation of tube vessels. These effects were reversed by an inhibitor (NS-398). In vivo, Diclofenac, a COX-2 inhibitor, decreased angiogenesis in mice presenting a colorectal cancer (Seed et al., 1997 May 1). Likewise, in a murine model of breast cancer, celecoxib (a selective COX-2 inhibitor) reduced metastasis and tumor burden through a decrease of micro vessel density and VEGF (Yoshinaka et al., 2006 Dec, Zhang et al., 2004 Sep). In clinical studies, patients with inflammatory breast cancers have increased levels of genes involved in angiogenesis such as VEGF (Van der Auwera et al., 2004 Dec 1). Patients with an inflammatory breast cancer benefit the most from anti-angiogenic treatment bevacizumab (Pierga et al., 2012 Apr).
The evidence was classified as “moderate” due to the lack of dose response studies.
KER 1266: Activation of the AhR leads to an increased endothelial migration
The activation of the AhR can lead to an increased endothelial cell migration. This was found when HMEC-1 or EA.hy926 cells were co-cultured with ER-positive MCF-7 cells and triple negative MDA-MB-231 cells (Pontillo et al., 2015 Nov 19, Zárate et al., 2020 Aug). The assay mainly used was the Matrigel® / tube formation assay. Only one study found an increase in endothelial cell proliferation and not migration, therefore it was not kept as a KE (Pontillo et al., 2015 Nov 19). The main pathway explaining this relationship was again related to the activation of COX2 and subsequently to the increase in VEGF. The association between the activation of the AhR and endothelial cell migration was classified as “weak” since only 2 studies explored this feature, and both used hexachlorobenzene as a stressor. However, these works were robust with strong evidence, and both found a reversed association after AhR blockage. No contradicting results were found in the scientific literature.
As opposed to our work, another AOP displayed a link between AhR activation and angiogenesis (AOP 150) and found that activation of the receptor could decrease VEGF production with moderate evidence and quantitative understanding. It must be noted that these AOPs applied only to chicken, zebrafish, and certain rodents whereas our AOP concerns humans. As detailed further, the AhR presents a variability between species which must be considered.
KER 1267: Increased endothelial migration promotes angiogenesis
Pontillo et al. treated mice with increasing doses of hexachlorobenzene and then calculated the vessel density in mammary fat pads (Pontillo et al., 2015 Nov 19). They found that mice treated with hexachlorobenzene had a higher vessel density with a dose–response concordance. Treatment by AhR antagonists completely reversed this association (Pontillo et al., 2015 Nov 19, Zárate et al., 2020 Aug). The relationship between endothelial migration and angiogenesis was not detailed here since there is existing extensive knowledge (Lamalice et al., 2007 Mar 30, Norton and Popel, 2016 Nov 14, Ausprunk and Folkman, 1977 Jul 1). The KER 12 was considered as “strong”.
KER 3137, 3138 and 3137: Increased tumor growth, increased invasion, and increased angiogenesis lead to breast cancer metastasis
Due to extensive data in the scientific literature and the empirical evidence in favor of these KERs, these KERs were not detailed here.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
2. Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer. 2020 May 15;126(10):2225–49.
3. Koual M, Tomkiewicz C, Cano-Sancho G, Antignac JP, Bats AS, Coumoul X. Environmental chemicals, breast cancer progression and drug resistance. Environ Health Glob Access Sci Source. 2020 Nov 17;19(1):117.
4. Koual M, Cano-Sancho G, Bats AS, Tomkiewicz C, Kaddouch-Amar Y, Douay-Hauser N, et al. Associations between persistent organic pollutants and risk of breast cancer metastasis. Environ Int. 2019;132:105028.
5. Koual M, Tomkiewicz C, Guerrera IC, Sherr D, Barouki R, Coumoul X. Aggressiveness and Metastatic Potential of Breast Cancer Cells Co-Cultured with Preadipocytes and Exposed to an Environmental Pollutant Dioxin: An in Vitro and in Vivo Zebrafish Study. Environ Health Perspect. 2021 Mar;129(3):37002.
6. Larigot L, Benoit L, Koual M, Tomkiewicz C, Barouki R, Coumoul X. Aryl Hydrocarbon Receptor and Its Diverse Ligands and Functions: An Exposome Receptor. Annu Rev Pharmacol Toxicol. 2021 Sep 9;
7. Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008 Feb;118(2):640–50.
8. Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene. 2000 Nov 16;19(48):5498–506.
9. Li ZD, Wang K, Yang XW, Zhuang ZG, Wang JJ, Tong XW. Expression of aryl hydrocarbon receptor in relation to p53 status and clinicopathological parameters in breast cancer. Int J Clin Exp Pathol. 2014;7(11):7931–7.
10. Zhao S, Ohara S, Kanno Y, Midorikawa Y, Nakayama M, Makimura M, et al. HER2 overexpression-mediated inflammatory signaling enhances mammosphere formation through up-regulation of aryl hydrocarbon receptor transcription. Cancer Lett. 2013 Mar 1;330(1):41–8.
11. Goode GD, Ballard BR, Manning HC, Freeman ML, Kang Y, Eltom SE. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int J Cancer. 2013 Dec 15;133(12):2769–80.
12. Kanno Y, Takane Y, Izawa T, Nakahama T, Inouye Y. The inhibitory effect of aryl hydrocarbon receptor repressor (AhRR) on the growth of human breast cancer MCF-7 cells. Biol Pharm Bull. 2006 Jun;29(6):1254–7.
13. Stanford EA, Wang Z, Novikov O, Mulas F, Landesman-Bollag E, Monti S, et al. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016 Mar 16;14:20.
14. Goode G, Pratap S, Eltom SE. Depletion of the aryl hydrocarbon receptor in MDA-MB-231 human breast cancer cells altered the expression of genes in key regulatory pathways of cancer. PloS One. 2014;9(6):e100103.
15. Safe S, Wormke M, Samudio I. Mechanisms of inhibitory aryl hydrocarbon receptor-estrogen receptor crosstalk in human breast cancer cells. J Mammary Gland Biol Neoplasia. 2000 Jul;5(3):295–306.
16. Carvaillo JC, Barouki R, Coumoul X, Audouze K. Linking Bisphenol S to Adverse Outcome Pathways Using a Combined Text Mining and Systems Biology Approach. Environ Health Perspect. 2019 Apr;127(4):47005.
17. Zgheib E, Kim MJ, Jornod F, Bernal K, Tomkiewicz C, Bortoli S, et al. Identification of non-validated endocrine disrupting chemical characterization methods by screening of the literature using artificial intelligence and by database exploration. Environ Int. 2021 Sep;154:106574.
18. Rugard M, Coumoul X, Carvaillo JC, Barouki R, Audouze K. Deciphering Adverse Outcome Pathway Network Linked to Bisphenol F Using Text Mining and Systems Toxicology Approaches. Toxicol Sci Off J Soc Toxicol. 2020 Jan 1;173(1):32–40.
19. Jornod F, Rugard M, Tamisier L, Coumoul X, Andersen HR, Barouki R, et al. AOP4EUpest: mapping of pesticides in adverse outcome pathways using a text mining tool. Bioinforma Oxf Engl. 2020 Aug 1;36(15):4379–81.
20. Jornod F, Jaylet T, Blaha L, Sarigiannis D, Tamisier L, Audouze K. AOP-helpFinder webserver: a tool for comprehensive analysis of the literature to support adverse outcome pathways development. Bioinforma Oxf Engl. 2021 Oct 30;btab750.
21. Wei CH, Kao HY, Lu Z. PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Res. 2013 Jul;41(Web Server issue):W518-522.
22. Wei CH, Allot A, Leaman R, Lu Z. PubTator central: automated concept annotation for biomedical full text articles. Nucleic Acids Res. 2019 Jul 2;47(W1):W587–93.
23. OECD. Users’ Handbook supplement to the Guidance Document for developing and assessing Adverse Outcome Pathways. 2018 Feb 14 [cited 2021 Sep 14]; Available from: https://www.oecd-ilibrary.org/environment/users-handbook-supplement-to-the-guidance-document-for-developing-and-assessing-adverse-outcome-pathways_5jlv1m9d1g32-en
24. Hill AB. The Environment and Disease: Association or Causation? Proc R Soc Med. 1965 May;58(5):295–300.
25. Vinken M, Landesmann B, Goumenou M, Vinken S, Shah I, Jaeschke H, et al. Development of an adverse outcome pathway from drug-mediated bile salt export pump inhibition to cholestatic liver injury. Toxicol Sci Off J Soc Toxicol. 2013 Nov;136(1):97–106.
26. Becker RA, Ankley GT, Edwards SW, Kennedy SW, Linkov I, Meek B, et al. Increasing Scientific Confidence in Adverse Outcome Pathways: Application of Tailored Bradford-Hill Considerations for Evaluating Weight of Evidence. Regul Toxicol Pharmacol. 2015 Aug 1;72(3):514–37.
27. Al-Dhfyan A, Alhoshani A, Korashy HM. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol Cancer. 2017 Jan 19;16(1):14.
28. Bekki K, Vogel H, Li W, Ito T, Sweeney C, Haarmann-Stemmann T, et al. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic Biochem Physiol. 2015 May;120:5–13.
29. Regan Anderson TM, Ma S, Perez Kerkvliet C, Peng Y, Helle TM, Krutilina RI, et al. Taxol Induces Brk-dependent Prosurvival Phenotypes in TNBC Cells through an AhR/GR/HIF-driven Signaling Axis. Mol Cancer Res MCR. 2018 Nov;16(11):1761–72.
30. Fujisawa Y, Li W, Wu D, Wong P, Vogel C, Dong B, et al. Ligand-independent activation of the arylhydrocarbon receptor by ETK (Bmx) tyrosine kinase helps MCF10AT1 breast cancer cells to survive in an apoptosis-inducing environment. Biol Chem. 2011 Oct;392(10):897–908.
31. Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011 Oct 15;71(20):6360–70.
32. Moran TJ, Gray S, Mikosz CA, Conzen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 2000 Feb 15;60(4):867–72.
33. Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004 Mar 1;64(5):1757–64.
34. Li Z, Dong J, Zou T, Du C, Li S, Chen C, et al. Dexamethasone induces docetaxel and cisplatin resistance partially through up-regulating Krüppel-like factor 5 in triple-negative breast cancer. Oncotarget. 2017 Feb 14;8(7):11555–65.
35. Li X, Lu Y, Liang K, Hsu JM, Albarracin C, Mills GB, et al. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene. 2012 Oct 4;31(40):4372–83.
36. Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995 Nov 3;83(3):493–501.
37. Vogel CFA, Li W, Sciullo E, Newman J, Hammock B, Reader JR, et al. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol. 2007 Nov;171(5):1538–48.
38. Thomas E, Dragojevic S, Price A, Raucher D. Thermally Targeted p50 Peptide Inhibits Proliferation and Induces Apoptosis of Breast Cancer Cell Lines. Macromol Biosci. 2020 Oct;20(10):e2000170.
39. Baud V, Jacque E. [The alternative NF-kB activation pathway and cancer: friend or foe?]. Med Sci MS. 2008 Dec;24(12):1083–8.
40. Demicco EG, Kavanagh KT, Romieu-Mourez R, Wang X, Shin SR, Landesman-Bollag E, et al. RelB/p52 NF-kappaB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IkappaB-alpha expression and promote carcinogenesis of the mammary gland. Mol Cell Biol. 2005 Nov;25(22):10136–47.
41. Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol. 2007 Apr;9(4):470–8.
42. Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001 May 25;276(21):18563–9.
43. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–74.
44. Parks AJ, Pollastri MP, Hahn ME, Stanford EA, Novikov O, Franks DG, et al. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol Pharmacol. 2014 Nov;86(5):593–608.
45. Pontillo CA, García MA, Peña D, Cocca C, Chiappini F, Alvarez L, et al. Activation of c-Src/HER1/STAT5b and HER1/ERK1/2 signaling pathways and cell migration by hexachlorobenzene in MDA-MB-231 human breast cancer cell line. Toxicol Sci Off J Soc Toxicol. 2011 Apr;120(2):284–96.
46. Qin XY, Wei F, Yoshinaga J, Yonemoto J, Tanokura M, Sone H. siRNA-mediated knockdown of aryl hydrocarbon receptor nuclear translocator 2 affects hypoxia-inducible factor-1 regulatory signaling and metabolism in human breast cancer cells. FEBS Lett. 2011 Oct 20;585(20):3310–5.
47. Nguyen CH, Brenner S, Huttary N, Atanasov AG, Dirsch VM, Chatuphonprasert W, et al. AHR/CYP1A1 interplay triggers lymphatic barrier breaching in breast cancer spheroids by inducing 12(S)-HETE synthesis. Hum Mol Genet. 2016 Nov 15;25(22):5006–16.
48. Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol Pharmacol. 2016 Nov;90(5):674–88.
49. Miret N, Pontillo C, Ventura C, Carozzo A, Chiappini F, Kleiman de Pisarev D, et al. Hexachlorobenzene modulates the crosstalk between the aryl hydrocarbon receptor and transforming growth factor-β1 signaling, enhancing human breast cancer cell migration and invasion. Toxicology. 2016 Jul 29;366–367:20–31.
50. Shan A, Leng L, Li J, Luo XM, Fan YJ, Yang Q, et al. TCDD-induced antagonism of MEHP-mediated migration and invasion partly involves aryl hydrocarbon receptor in MCF7 breast cancer cells. J Hazard Mater. 2020 Nov 5;398:122869.
51. Dwyer AR, Kerkvliet CP, Krutilina RI, Playa HC, Parke DN, Thomas WA, et al. Breast Tumor Kinase (Brk/PTK6) Mediates Advanced Cancer Phenotypes via SH2-Domain Dependent Activation of RhoA and Aryl Hydrocarbon Receptor (AhR) Signaling. Mol Cancer Res MCR. 2021 Feb;19(2):329–45.
52. Narasimhan S, Stanford Zulick E, Novikov O, Parks AJ, Schlezinger JJ, Wang Z, et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. Int J Mol Sci. 2018 May 7;19(5):1388.
53. Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Lee JN, Chai CY, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J Off Publ Fed Am Soc Exp Biol. 2012 Feb;26(2):778–87.
54. Pontillo CA, Rojas P, Chiappini F, Sequeira G, Cocca C, Crocci M, et al. Action of hexachlorobenzene on tumor growth and metastasis in different experimental models. Toxicol Appl Pharmacol. 2013 May 1;268(3):331–42.
55. Miller ME, Holloway AC, Foster WG. Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin Exp Metastasis. 2005;22(2):149–56.
56. Belguise K, Guo S, Yang S, Rogers AE, Seldin DC, Sherr DH, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007 Dec 15;67(24):11742–50.
57. Yamashita N, Saito N, Zhao S, Terai K, Hiruta N, Park Y, et al. Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp Cell Res. 2018 May 1;366(1):34–40.
58. Miret N, Zappia CD, Altamirano G, Pontillo C, Zárate L, Gómez A, et al. AhR ligands reactivate LINE-1 retrotransposon in triple-negative breast cancer cells MDA-MB-231 and non-tumorigenic mammary epithelial cells NMuMG. Biochem Pharmacol. 2020 May;175:113904.
59. Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3’-diindolylmethane in breast cancer cells. J Nutr. 2009 Jan;139(1):26–32.
60. Vogel CFA, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, et al. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys. 2011 Aug 1;512(1):78–86.
61. Kolasa E, Houlbert N, Balaguer P, Fardel O. AhR- and NF-κB-dependent induction of interleukin-6 by co-exposure to the environmental contaminant benzanthracene and the cytokine tumor necrosis factor-α in human mammary MCF-7 cells. Chem Biol Interact. 2013 Apr 25;203(2):391–400.
62. Vacher S, Castagnet P, Chemlali W, Lallemand F, Meseure D, Pocard M, et al. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PloS One. 2018;13(1):e0190619.
63. Malik DES, David RM, Gooderham NJ. Interleukin-6 selectively induces drug metabolism to potentiate the genotoxicity of dietary carcinogens in mammary cells. Arch Toxicol. 2019 Oct;93(10):3005–20.
64. Jönsson ME, Kubota A, Timme-Laragy AR, Woodin B, Stegeman JJ. Ahr2-dependence of PCB126 effects on the swim bladder in relation to expression of CYP1 and cox-2 genes in developing zebrafish. Toxicol Appl Pharmacol. 2012 Dec 1;265(2):166–74.
65. Degner SC, Kemp MQ, Hockings JK, Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer. 2007;59(2):248–57.
66. Gilhooly EM, Rose DP. The association between a mutated ras gene and cyclooxygenase-2 expression in human breast cancer cell lines. Int J Oncol. 1999 Aug;15(2):267–70.
67. Liu XH, Rose DP. Differential expression and regulation of cyclooxygenase-1 and -2 in two human breast cancer cell lines. Cancer Res. 1996 Nov 15;56(22):5125–7.
68. Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002 Feb 1;62(3):632–5.
69. Parrett M, Harris R, Joarder F, Ross M, Clausen K, Robertson F. Cyclooxygenase-2 gene expression in human breast cancer. Int J Oncol. 1997 Mar;10(3):503–7.
70. Singh B, Berry JA, Shoher A, Ramakrishnan V, Lucci A. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol. 2005 May;26(5):1393–9.
71. Harris RE, Alshafie GA, Abou-Issa H, Seibert K. Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Res. 2000 Apr 15;60(8):2101–3.
72. Schlichting JA, Soliman AS, Schairer C, Schottenfeld D, Merajver SD. Inflammatory and non-inflammatory breast cancer survival by socioeconomic position in the Surveillance, Epidemiology, and End Results database, 1990-2008. Breast Cancer Res Treat. 2012 Aug;134(3):1257–68.
73. Fouad TM, Barrera AMG, Reuben JM, Lucci A, Woodward WA, Stauder MC, et al. Inflammatory breast cancer: a proposed conceptual shift in the UICC-AJCC TNM staging system. Lancet Oncol. 2017 Apr;18(4):e228–32.
74. Takahashi Y, Kawahara F, Noguchi M, Miwa K, Sato H, Seiki M, et al. Activation of matrix metalloproteinase-2 in human breast cancer cells overexpressing cyclooxygenase-1 or -2. FEBS Lett. 1999 Oct 22;460(1):145–8.
75. Sivula A, Talvensaari-Mattila A, Lundin J, Joensuu H, Haglund C, Ristimäki A, et al. Association of cyclooxygenase-2 and matrix metalloproteinase-2 expression in human breast cancer. Breast Cancer Res Treat. 2005 Feb;89(3):215–20.
76. Larkins TL, Nowell M, Singh S, Sanford GL. Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer. 2006 Jul 10;6:181.
77. Guyton DP, Evans DM, Sloan-Stakleff KD. Urokinase Plasminogen Activator Receptor (uPAR): A Potential Indicator of Invasion for In Situ Breast Cancer. Breast J. 2000 Mar;6(2):130–6.
78. Pontillo C, Español A, Chiappini F, Miret N, Cocca C, Alvarez L, et al. Hexachlorobenzene promotes angiogenesis in vivo, in a breast cancer model and neovasculogenesis in vitro, in the human microvascular endothelial cell line HMEC-1. Toxicol Lett. 2015 Nov 19;239(1):53–64.
79. Zárate LV, Pontillo CA, Español A, Miret NV, Chiappini F, Cocca C, et al. Angiogenesis signaling in breast cancer models is induced by hexachlorobenzene and chlorpyrifos, pesticide ligands of the aryl hydrocarbon receptor. Toxicol Appl Pharmacol. 2020 Aug 15;401:115093.
80. Harris RE, Casto BC, Harris ZM. Cyclooxygenase-2 and the inflammogenesis of breast cancer. World J Clin Oncol. 2014 Oct 10;5(4):677–92.
81. Kirkpatrick K, Ogunkolade W, Elkak A, Bustin S, Jenkins P, Ghilchik M, et al. The mRNA expression of cyclo-oxygenase-2 (COX-2) and vascular endothelial growth factor (VEGF) in human breast cancer. Curr Med Res Opin. 2002;18(4):237–41.
82. Costa C, Soares R, Reis-Filho JS, Leitão D, Amendoeira I, Schmitt FC. Cyclo-oxygenase 2 expression is associated with angiogenesis and lymph node metastasis in human breast cancer. J Clin Pathol. 2002 Jun;55(6):429–34.
83. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998 May 29;93(5):705–16.
84. Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2000 Jan;6(1):135–8.
85. Seed MP, Brown JR, Freemantle CN, Papworth JL, Colville-Nash PR, Willis D, et al. The inhibition of colon-26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Res. 1997 May 1;57(9):1625–9.
86. Yoshinaka R, Shibata MA, Morimoto J, Tanigawa N, Otsuki Y. COX-2 inhibitor celecoxib suppresses tumor growth and lung metastasis of a murine mammary cancer. Anticancer Res. 2006 Dec;26(6B):4245–54.
87. Zhang S, Lawson KA, Simmons-Menchaca M, Sun L, Sanders BG, Kline K. Vitamin E analog alpha-TEA and celecoxib alone and together reduce human MDA-MB-435-FL-GFP breast cancer burden and metastasis in nude mice. Breast Cancer Res Treat. 2004 Sep;87(2):111–21.
88. Van der Auwera I, Van Laere SJ, Van den Eynden GG, Benoy I, van Dam P, Colpaert CG, et al. Increased angiogenesis and lymphangiogenesis in inflammatory versus noninflammatory breast cancer by real-time reverse transcriptase-PCR gene expression quantification. Clin Cancer Res Off J Am Assoc Cancer Res. 2004 Dec 1;10(23):7965–71.
89. Pierga JY, Petit T, Delozier T, Ferrero JM, Campone M, Gligorov J, et al. Neoadjuvant bevacizumab, trastuzumab, and chemotherapy for primary inflammatory HER2-positive breast cancer (BEVERLY-2): an open-label, single-arm phase 2 study. Lancet Oncol. 2012 Apr;13(4):375–84.
90. Lamalice L, Le Boeuf F, Huot J. Endothelial Cell Migration During Angiogenesis. Circ Res. 2007 Mar 30;100(6):782–94.
91. Norton KA, Popel AS. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci Rep. 2016 Nov 14;6(1):36992.
92. Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977 Jul 1;14(1):53–65.
93. Hahn ME. Aryl hydrocarbon receptors: diversity and evolution. Chem Biol Interact. 2002 Sep 20;141(1–2):131–60.
94. Korkalainen M, Tuomisto J, Pohjanvirta R. The AH receptor of the most dioxin-sensitive species, guinea pig, is highly homologous to the human AH receptor. Biochem Biophys Res Commun. 2001 Aug 3;285(5):1121–9.
95. Cohen-Barnhouse AM, Zwiernik MJ, Link JE, Fitzgerald SD, Kennedy SW, Hervé JC, et al. Sensitivity of Japanese quail (Coturnix japonica), Common pheasant (Phasianus colchicus), and White Leghorn chicken (Gallus gallus domesticus) embryos to in ovo exposure to TCDD, PeCDF, and TCDF. Toxicol Sci Off J Soc Toxicol. 2011 Jan;119(1):93–103.
96. Doering JA, Giesy JP, Wiseman S, Hecker M. Predicting the sensitivity of fishes to dioxin-like compounds: possible role of the aryl hydrocarbon receptor (AhR) ligand binding domain. Environ Sci Pollut Res Int. 2013 Mar;20(3):1219–24.
97. DiNatale BC, Schroeder JC, Francey LJ, Kusnadi A, Perdew GH. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J Biol Chem. 2010 Aug 6;285(32):24388–97.
98. Liu Z, Wu X, Zhang F, Han L, Bao G, He X, et al. AhR expression is increased in hepatocellular carcinoma. J Mol Histol. 2013 Aug;44(4):455–61.
99. Stanford EA, Ramirez-Cardenas A, Wang Z, Novikov O, Alamoud K, Koutrakis P, et al. Role for the Aryl Hydrocarbon Receptor and Diverse Ligands in Oral Squamous Cell Carcinoma Migration and Tumorigenesis. Mol Cancer Res MCR. 2016 Aug;14(8):696–706.
100. Koliopanos A, Kleeff J, Xiao Y, Safe S, Zimmermann A, Büchler MW, et al. Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene. 2002 Sep 5;21(39):6059–70.
101. Chang JT, Chang H, Chen PH, Lin SL, Lin P. Requirement of aryl hydrocarbon receptor overexpression for CYP1B1 up-regulation and cell growth in human lung adenocarcinomas. Clin Cancer Res Off J Am Assoc Cancer Res. 2007 Jan 1;13(1):38–45.
102. Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, et al. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004 Jul 15;64(14):4707–10.
103. Ishida M, Mikami S, Shinojima T, Kosaka T, Mizuno R, Kikuchi E, et al. Activation of aryl hydrocarbon receptor promotes invasion of clear cell renal cell carcinoma and is associated with poor prognosis and cigarette smoke. Int J Cancer. 2015 Jul 15;137(2):299–310.
104. Ishida M, Mikami S, Kikuchi E, Kosaka T, Miyajima A, Nakagawa K, et al. Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by upregulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. Carcinogenesis. 2010 Feb;31(2):287–95.
105. Diry M, Tomkiewicz C, Koehle C, Coumoul X, Bock KW, Barouki R, et al. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene. 2006 Sep 7;25(40):5570–4.
106. John K, Lahoti TS, Wagner K, Hughes JM, Perdew GH. The Ah receptor regulates growth factor expression in head and neck squamous cell carcinoma cell lines. Mol Carcinog. 2014 Oct;53(10):765–76.
107. AOP-Wiki [Internet]. [cited 2021 Sep 15]. Available from: https://aopwiki.org/
108. Brooks J, Eltom SE. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr Cancer Drug Targets. 2011 Jun;11(5):654–69.
109. Currier N, Solomon SE, Demicco EG, Chang DLF, Farago M, Ying H, et al. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol Pathol. 2005;33(6):726–37.
110. Pirkle JL, Wolfe WH, Patterson DG, Needham LL, Michalek JE, Miner JC, et al. Estimates of the half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Vietnam Veterans of Operation Ranch Hand. J Toxicol Environ Health. 1989;27(2):165–71.
111. Pesatori AC, Consonni D, Rubagotti M, Grillo P, Bertazzi PA. Cancer incidence in the population exposed to dioxin after the “Seveso accident”: twenty years of follow-up. Environ Health Glob Access Sci Source. 2009 Sep 15;8:39.
112. Warner M, Eskenazi B, Mocarelli P, Gerthoux PM, Samuels S, Needham L, et al. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s Health Study. Environ Health Perspect. 2002 Jul;110(7):625–8.
Fulda, S., & Debatin, K. M. (2007). Apoptosis signaling in cancer. Experimental Cell Research, 313(9), 1503-1515. https://pubmed.ncbi.nlm.nih.gov/17296041/
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446472/
Luo, X., & Heng, H. H. (2003). Apoptosis and cancer therapy: lessons from the past and new directions. Current Pharmaceutical Design, 9(21), 1803-1816. https://pubmed.ncbi.nlm.nih.gov/14574468/
Schmitt, C. A., et al. (2000). Senescence and apoptosis. Oncogene, 19(56), 6207-6210. https://pubmed.ncbi.nlm.nih.gov/11101894/
Benson, A. B., et al. (2008). Tumor size and stage in colon and rectal cancer: Consistent prognostic factors over time. Annals of Surgery, 247(3), 421-429.
Clark, B. A., et al. (2009). New insights into the biology of human malignant melanoma. Annual Review of Medicine, 60, 361-377. https://pubmed.ncbi.nlm.nih.gov/19021562/
Friedl, P., & Weigelin, B. (2008. Interstitial cell migration and invasion in cancer and metastasis. Nature Reviews Cancer, 8(10), 700-710. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2724717/
Fearon, E. R., & Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell, 61(5), 759-767. https://pubmed.ncbi.nlm.nih.gov/2202721/
Olive, P. L., et al. (2009. Inhibition of Hedgehog signaling enhances sensitivity of pancreatic cancer cells to gemcitabine. Clinical Cancer Research, 15(8), 2553-2562. https://pubmed.ncbi.nlm.nih.gov/19318265/
Appendix 1
List of MIEs in this AOP
Event: 18: Activation, AhR
Short Name: Activation, AhR
Key Event Component
| Process | Object | Action |
|---|---|---|
| aryl hydrocarbon receptor activity | aryl hydrocarbon receptor | increased |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Benzidine |
| Dibenzo-p-dioxin |
| Polychlorinated biphenyl |
| Polychlorinated dibenzofurans |
| Hexachlorobenzene |
| Polycyclic aromatic hydrocarbons (PAHs) |
Biological Context
| Level of Biological Organization |
|---|
| Molecular |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| zebra danio | Danio rerio | High | NCBI |
| Gallus gallus | Gallus gallus | High | NCBI |
| Pagrus major | Pagrus major | High | NCBI |
| Acipenser transmontanus | Acipenser transmontanus | High | NCBI |
| Acipenser fulvescens | Acipenser fulvescens | High | NCBI |
| rainbow trout | Oncorhynchus mykiss | High | NCBI |
| Salmo salar | Salmo salar | High | NCBI |
| Xenopus laevis | Xenopus laevis | High | NCBI |
| Ambystoma mexicanum | Ambystoma mexicanum | High | NCBI |
| Phasianus colchicus | Phasianus colchicus | High | NCBI |
| Coturnix japonica | Coturnix japonica | High | NCBI |
| mouse | Mus musculus | High | NCBI |
| rat | Rattus norvegicus | High | NCBI |
| human | Homo sapiens | High | NCBI |
| Microgadus tomcod | Microgadus tomcod | High | NCBI |
| Homo sapiens | Homo sapiens | NCBI |
| Life Stage | Evidence |
|---|---|
| Embryo | High |
| Development | High |
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
The AHR structure has been shown to contribute to differences in species sensitivity to DLCs in several animal models. In 1976, a 10-fold difference was reported between two strains of mice (non-responsive DBA/2 mouse, and responsive C57BL/6 14 mouse) in CYP1A induction, lethality and teratogenicity following TCDD exposure[3]. This difference in dioxin sensitivity was later attributed to a single nucleotide polymorphism at position 375 (the equivalent position of amino acid residue 380 in chicken) in the AHR LBD[30][19][31]. Several other studies reported the importance of this amino acid in birds and mammals[32][30][22][33][34][35][31][36]. It has also been shown that the amino acid at position 319 (equivalent to 324 in chicken) plays an important role in ligand-binding affinity to the AHR and transactivation ability of the AHR, due to its involvement in LBD cavity volume and its steric effect[35]. Mutation at position 319 in the mouse eliminated AHR DNA binding[35].
The first study that attempted to elucidate the role of avian AHR1 domains and key amino acids within avian AHR1 in avian differential sensitivity was performed by Karchner et al.[22]. Using chimeric AHR1 constructs combining three AHR1 domains (DBD, LBD and TAD) from the chicken (highly sensitive to DLC toxicity) and common tern (resistant to DLC toxicity), Karchner and colleagues[22], showed that amino acid differences within the LBD were responsible for differences in TCDD sensitivity between the chicken and common tern. More specifically, the amino acid residues found at positions 324 and 380 in the AHR1 LBD were associated with differences in TCDD binding affinity and transactivation between the chicken (Ile324_Ser380) and common tern (Val324_Ala380) receptors[22]. Since the Karchner et al. (2006) study was conducted, the predicted AHR1 LBD amino acid sequences were been obtained for over 85 species of birds and 6 amino acid residues differed among species[14][37] . However, only the amino acids at positions 324 and 380 in the AHR1 LBD were associated with differences in DLC toxicity in ovo and AHR1-mediated gene expression in vitro[14][37][16]. These results indicate that avian species can be divided into one of three AHR1 types based on the amino acids found at positions 324 and 380 of the AHR1 LBD: type 1 (Ile324_Ser380), type 2 (Ile324_Ala380) and type 3 (Val324_Ala380)[14][37][16].
- Little is known about differences in binding affinity of AhRs and how this relates to sensitivity in non-avian taxa.
- Low binding affinity for DLCs of AhR1s of African clawed frog (Xenopus laevis) and axolotl (Ambystoma mexicanum) has been suggested as a mechanism for tolerance of these amphibians to DLCs (Lavine et al 2005; Shoots et al 2015).
- Among reptiles, only AhRs of American alligator (Alligator mississippiensis) have been investigated and little is known about the sensitivity of American alligator or other reptiles to DLCs (Oka et al 2016).
- Among fishes, great differences in sensitivity to DLCs are known both for AhRs and for embryos among species that have been tested (Doering et al 2013; 2014).
- Differences in binding affinity of the AhR2 have been demonstrated to explain differences in sensitivity to DLCs between sensitive and tolerant populations of Atlantic Tomcod (Microgadus tomcod) (Wirgin et al 2011).
- This was attributed to the rapid evolution of populations in highly contaminated areas of the Hudson River, resulting in a 6-base pair deletion in the AHR sequence (outside the LBD) and reduced ligand binding affinity, due to reduces AHR protein stability.
- Information is not yet available regarding whether differences in binding affinity of AhRs of fishes are predictive of differences in sensitivity of embryos, juveniles, or adults (Doering et al 2013).
The AhR is a very conserved and ancient protein (95) and the AhR is present in human and mice (96–98). The AhR is present in human physiology and pathology. The AhR is highly expressed at several important physiological barriers such as the placenta, lung, gastrointestinal system, and liver in human (Wakx, Marinelli, Watanabe). In these tissues, the AhR is involved in both detoxication processes involving xenobiotic metabolizing enzymes such as cytochromes P450, and in immune functions translating chemical signals into immune defence pathways (Marinelli, Stobbe). Moreover, it has a regulatory role in human dendritic cells and myelination (Kado, Shackleford). The lung constitutes another barrier exposed to components of air pollution such as particles and hydrocarbons (air pollution, cigarette smoke). The AhR detects such hydrocarbons and protects the pulmonary cells from their deleterious effects through metabolization. The regulatory effect on blood cells of the AhR, balancing different related cell types, can be extended to the megakaryocytes and their precursors; indeed, StemRegenin 1 (SR1), an antagonist of the AhR increases the human population of CD34+CD41low cells, a fraction of very efficient precursors of proplatelets (Bock). The occurrence of a nystagmus has been subsequently diagnosed in humans bearing a AhR mutation (Borovok).
In human cancer, the AhR has either a pro or con tumor effect depending on the tissue, the ligand, and the duration of the activation (Zudaire, Chang, Litzenburg, Gramatzki, Lin, Wang). In human breast cancer, the AhR is thoughts to be responsible of its progression (Goode, Kanno, Optiz, Novikov, Hall, Subramaniam, Barhoover). In human mammary benign cells, Brooks et al. noted that a high level of AhR was associated with a modified cell cycle (with a 50% increase in population doubling time in cells expressing the AhR by more than 3-fold) and EMT including increased cell migration. Narasimnhan et al. found that suppression of the AhR pathway had a pro-tumorigenic effect in vitro (EMT, tumor migration) in triple negative breast cancer.
Many endogenous and exogenous ligands are present for the AhR in human (Optiz, Adachi, Schroeder, Rothhammer). Indoles, such as indole-3-carbinol or one of its secondary metabolites, 3-3'- Diindolylmethane, are degradation products found in cruciferous vegetables and characterized as AhR ligands (Ema, Kall, Miller) they are also inducers of the human and rat CYP1A1 (Optiz). FICZ is the most potent AhR ligand known to date: it has a stronger affinity than TCDD for the human AhR (TCDD Kd=0.48 nM/FICZ Kd=0.07 nM) (Coumoul).
Key Event Description
The AHR Receptor
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that belongs to the basic helix-loop-helix Per-ARNT-Sim (bHLH-PAS) superfamily and consists of three domains: the DNA-binding domain (DBD), ligand binding domain (LBD) and transactivation domain (TAD)[1]. Other members of this superfamily include the AHR nuclear translocator (ARNT), which acts as a dimerization partner of the AHR [2][3]; Per, a circadian transcription factor; and Sim, the “single-minded” protein involved in neuronal development [4][5]. This group of proteins shares a highly conserved PAS domain and is involved in the detection of and adaptation to environmental change[4].
Investigations of invertebrates possessing early homologs of the AhR suggest that the AhR evolutionarily functioned in regulation of the cell cycle, cellular proliferation and differentiation, and cell-to-cell communications (Hahn et al 2002). However, critical functions in angiogenesis, regulation of the immune system, neuronal processes, metabolism, development of the heart and other organ systems, and detoxification have emerged sometime in early vertebrate evolution (Duncan et al., 1998; Emmons et al., 1999; Lahvis and Bradfield, 1998).
The molecular Initiating Event

The molecular mechanism for AHR-mediated activation of gene expression is presented in Figure 1. In its unliganded form, the AHR is part of a cytosolic complex containing heat shock protein 90 (HSP90), the HSP90 co-chaperone p23 and AHR-interacting protein (AIP)[6]. Upon ligand binding, the AHR migrates to the nucleus where it dissociates from the cytosolic complex and forms a heterodimer with ARNT[7]. The AHR-ARNT complex then binds to a xenobiotic response element (XRE) found in the promoter of an AHR-regulated gene and recruits co-regulators such as CREB binding protein/p300, steroid receptor co-activator (SRC) 1, SRC-2, SRC-3 and nuclear receptor interacting protein 1, leading to induction or repression of gene expression[6]. Expression levels of several genes, including phase I (e.g. cytochrome P450 (CYP) 1A, CYP1B, CYP2A) and phase II enzymes (e.g. uridine diphosphate glucuronosyl transferase (UDP-GT), glutathione S-transferases (GSTs)), as well as genes involved in cell proliferation (transforming growth factor-beta, interleukin-1 beta), cell cycle regulation (p27, jun-B) and apoptosis (Bax), are regulated through this mechanism [6][8][7][9].
AHR Isoforms
- Over time the AhR has undergone gene duplication and diversification in vertebrates, which has resulted in multiple clades of AhR, namely AhR1, AhR2, and AhR3 (Hahn 2002).
- Fishes and birds express AhR1s and AhR2s, while mammals express a single AhR that is homologous to the AhR1 (Hahn 2002; Hahn et al 2006).
- The AhR3 is poorly understood and known only from some cartilaginous fishes (Hahn 2002).
- Little is known about diversity of AhRs in reptiles and amphibians (Hahn et al 2002).
- In some taxa, subsequent genome duplication events have further led to multiple isoforms of AhRs in some species, with up to four isoforms of the AhR (α, β, δ, γ) having been identified in Atlantic salmon (Salmo salar) (Hansson et al 2004).
- Although homologs of the AhR have been identified in some invertebrates, compared to vertebrates these AhRs have differences in binding of ligands in the species investigated to date (Hahn 2002; Hahn et al 1994).
Roles of isoforms in birds:
Two AHR isoforms (AHR1 and AHR2) have been identified in the black-footed albatross (Phoebastria nigripes), great cormorant (Phalacrocorax carbo) and domestic chicken (Gallus gallus domesticus)[10]. AHR1 mRNA levels were similar in the kidney, heart, lung, spleen, brain, gonad and intestine from the great cormorant but were lower in muscle and pancreas. AHR2 expression was mainly observed in the liver, but was also detected in gonad, brain and intestine. AHR1 levels represented a greater proportion (80%) of total AHR levels than AHR2 in the cormorant liver[10], and while both AHR isoforms bound to TCDD, AHR2 was less effective at inducing TCDD-dependent transactivation compared to AHR1 in black-footed albatross, great cormorant and domestic chicken[11][10].
- AhR1 and AhR2 both bind and are activated by TCDD in vitro (Yasui et al 2007).
- AhR1 has greater binding affinity and sensitivity to activation by TCDD relative to AhR2 (Yasui et al 2007).
- AhR1 is believed to mediate toxicities of DLCs, while AhR2 has no known role in toxicities (Farmahin et al 2012; Farmahin et al 2013; Manning et al 2012).
Roles of isoforms in fishes:
- AhR1 and AhR2 both bind and are activated by TCDD in vitro (Bak et al 2013; Doering et al 2014; 2015; Karchner et al 1999; 2005).
- AhR1 has greater sensitivity to activation by TCDD than AhR2 in red seabream (Pagrus major), white sturgeon (Acipenser transmontanus), and lake sturgeon (Acipenser fulvescens) (Bak et al 2013; Doering et al 2014; 2015)
- AhR2 has greater binding affinity or activation by TCDD than AhR1 in zebrafish (Danio rerio) and mummichog (Fundulus heteroclitus) (Karchner et al 1999; 2005).
- AhR2 is believed to mediate toxicities in fishes, while AhR1 has no known role in toxicities. Specifically, knockdown of AhR2 protects against toxicities of dioxin-like compounds (DLCs) and polycyclic aromatic hydrocarbons (PAHs) in zebrafish (Danio rerio) and mummichog (Fundulus heteroclitus), while knockdown of AhR1 offers no protection (Clark et al 2010; Prasch et al 2003; Van Tiem & Di Giulio 2011).
Roles of isoforms in amphibians and reptiles:
- Less is known about AhRs of amphibians or reptiles.
- AhR1 is believed to mediate toxicities in amphibians (Hahn 2002; Lavine et al 2005; Oka et al 2016; Shoots et al 2015). However, all AhRs of amphibians that have been investigated have very low affinity for TCDD (Hahn 2002; Lavine et al 2005; Oka et al 2016; Shoots et al 2015).
- Both AhR1s and AhR2 of American alligator (Alligator mississippiensis) are activated by agonists with comparable sensitivities (Oka et al 2016). AhRs of no other reptiles have been investigated.
How it is Measured or Detected
Methods that have been previously reviewed and approved by a recognized authority should be included in the Overview section above. All other methods, including those well established in the published literature, should be described here. Consider the following criteria when describing each method: 1. Is the assay fit for purpose? 2. Is the assay directly or indirectly (i.e. a surrogate) related to a key event relevant to the final adverse effect in question? 3. Is the assay repeatable? 4. Is the assay reproducible?
Transactivation Reporter Gene Assays (recommended approach)
Transient transfection transactivation
Transient transfection transactivation is the most common method for evaluating nuclear receptor activation[12]. Full-length AHR cDNAs are cloned into an expression vector along with a reporter gene construct (chimeric luciferase, P-lactamase or CAT reporter vectors containing the appropriate response elements for the gene of interest). There are a number of commercially available cell lines that can serve as recipients for these vectors (CV-1, HuH7, FLC-7, LS174T, LS180 MCF-7, HEC1, LLC-PK1, HEK293, HepG2, and Caco-2 cells)[12]. The greatest advantage of using transfected cells, rather than primary cell cultures, is the assurance that the nuclear receptor of interest is responsible for the observed induction. This would not be possible in a primary cell culture due to the co-regulation of different receptors for the same target genes. This model makes it easy to compare the responsiveness of the AHR across multiple species under the same conditions simply by switching out the AHR clone. One disadvantage to the transient transfection assay is the inherent variability associated with transfection efficiency, leading to a movement towards the use of stable cell lines containing the nuclear receptor and reporter gene linked to the appropriate response elements[12].
Luciferase reporter gene (LRG) assay
The described luciferase reporter gene (LRG) assays have been used to investigate activation of AhRs of:
- Humans (Homo sapiens) (Abnet et al 1999)
- Species of birds, namely chicken (Gallus gallus), ring-necked pheasant (Phasianus colchicus), Japanese quail (Coturnix japonica), and common tern (Sterna hirundo) (Farmahin et al 2012; Manning et al 2013), Mutant AhR1s with ligand binding domains resembling those of at least 86 avian species have also been investigated (Farmahin et al 2013). AhR2s of birds have only been investigated in black-footed albatross (Phoebastria nigripes) and common cormorant (Phalacrocorax carbo) (Yasio et al 2007).
- American alligator (Alligator mississippiensis) is the only reptile for which AhR activation has been investigated (Oka et al 2016), AhR1A, AhR1B, and AhR2 of American alligator were assayed (Oka et al 2016).
- AhR1 of two amphibians have been investigated, namely African clawed frog (Xenopus laevis) and salamander (Ambystoma mexicanum) (Lavine et al 2005; Shoots et al 2015; Ohi et al 2003),
- AhR1s and AhR2s of several species of fish have been investigated, namely Atlantic salmon (Salmo salar), Atlantic tomcod (Microgadus tomcod), white sturgeon (Acipenser transmontanus), rainbow trout (Onchorhynchys mykiss), red seabream (Pagrus major), lake sturgeon (Acipenser fulvescens), and zebrafish (Danio rerio) (Andreasen et al 2002; Abnet et al 1999; Bak et al 2013; Doering et al 2014; 2015; Evans et al 2005; Hansson & Hahn 2008; Karchner et al 1999; Tanguay et al 1999; Wirgin et al 2011).
For demonstrative purposes, a luciferase reporter gene assay used to measure AHR1-mediated transactivation for avian species is described here. However, comparable assays are utilized for investigating AHR1s and AHR2s of all taxa. A monkey kidney cell line (Cos-7) that has low endogenous AHR1 expression was transfected with the appropriate avian AHR1 clone, cormorant ARNT1, a CYP1A5 firefly luciferase reporter construct and a Renilla luciferase vector to control for transfection efficiency. After seeding, the cells were exposed to DLC and luciferase activity was measured using a luminometer. Luminescence, which is proportional to the extent of AHR activation, is expressed as the ratio of firefly luciferase units to Renilla luciferase units [13]. This particular assay was modified from its original version to increase throughput efficiency; (a) cells were seeded in 96-well plates rather than Petri dishes or 48- well plates, (b) DLCs were added directly to the wells without changing the cell culture medium, and (c) the same 96-well plates were used to measure luminescence without lysing the cells and transferring to another plate. Similar reporter gene assays have been used to measure AHR1 activation in domestic and wild species of birds, including the chicken, ring-necked pheasant (Phasianus colchicus), Japanese quail (Coturnix japonica), great cormorant, black-footed albatross and peregrine falcon (Falco peregrinus).[14][13][15][11][16][17]
Transactivation in stable cell lines
Stable cell lines have been developed and purified to the extent that each cell contains both the nuclear receptor and appropriate reporter vector, eliminating the variability associated with transfection [12]. A stable human cell line containing a luciferase reporter driven by multiple dioxin response elements has been developed that is useful in identifying AhR agonists and antagonists[18]. An added benefit of this model is the potential to multiplex 3 assays in a single well: receptor activation, cell viability and enzyme activity[12]. Such assays are used extensively in drug discovery due to their high throughput efficiency, and may serve just as useful for risk assessment purposes.
Ligand-Binding Assays
Ligand binding assays measure the ability of a test compound to compete with a labeled, high-affinity reference ligand for the LBD of a nuclear receptor. It is important to note that ligand binding does not necessitate receptor activation and therefore cannot distinguish between agonists and antagonists; however, binding affinities of AHR ligands are highly correlated with chemical potencies[19] and can explain differences in species sensitivities to DLCs[20][21][22]; they are therefore worth mentioning. Binding affinity and efficacy have been used to develop structure-activity relationships for AHR disruption[20][23] that are potentially useful in risk-assessment. There has been tremendous progress in the development of ligand-binding assays for nuclear receptors that use homogenous assay formats (no wash steps) allowing for the detection of low-affinity ligands, many of which do not require a radiolabel and are amenable to high throughput screening[24][12]. This author however was unable to find specific examples of such assays in the context of AHR binding and therefore some classic radioligand assays are described instead.
Hydroxyapatite (HAP) binding assay
The HAP binding assay makes use of an in vitro transcription/translation method to synthesize the AHR protein, which is then incubated with radiolabeled TDCPP and a HAP pellet. The occupied protein adsorbs to the HAP and the radioactivity is measured to determine saturation binding. An additional ligand can also be included in the mixture in order to determine its binding affinity relative to TCDD (competitive binding)[25][22]. This assay is simple, repeatable and reproducible; however, it is insensitive to weak ligand-receptor interactions[22][21][26].
Whole cell filtration binding assay
Dold and Greenlee[27] developed a method to detect specific binding of TCDD to whole mammalian cells in culture and was later modified by Farmahin et al.[21] for avian species. The cultured cells are incubated with radiolabeled TCDD with or without the presence of a competing ligand and filtered. The occupied protein adsorbs onto the filter and the radioactivity is measured to determine saturation binging and/or competitive binding. This assay is able to detect weak ligand-receptor interactions that are below the detection limit of the HAP assay[21].
Protein-DNA Interaction Assays
The active AHR complexed with ARNT can be measured using protein-DNA interaction assays. Two methods are described in detail by Perez-Romero and Imperiale[28]. Chromatin immunoprecipitation measures the interaction of proteins with specific genomic regions in vivo. It involves the treatment of cells with formaldehyde to crosslink neighboring protein-protein and protein-DNA molecules. Nuclear fractions are isolated, the genomic DNA is sheared, and nuclear lysates are used in immunoprecipitations with an antibody against the protein of interest. After reversal of the crosslinking, the associated DNA fragments are sequenced. Enrichment of specific DNA sequences represents regions on the genome that the protein of interest is associated with in vivo. Electrophoretic mobility shift assay (EMSA) provides a rapid method to study DNA-binding protein interactions in vitro. This relies on the fact that complexes of protein and DNA migrate through a nondenaturing polyacrylamide gel more slowly than free DNA fragments. The protein-DNA complex components are then identified with appropriate antibodies. The EMSA assay was found to be consistent with the LRG assay in chicken hepatoma cells dosed with dioxin-like compounds[29].
In silico Approaches
In silico homology modeling of the ligand binding domain of the AHR in combination with molecular docking simulations can provide valuable insight into the transactivation-potential of a diverse array of AHR ligands. Such models have been developed for multiple AHR isoforms and ligands (high/low affinity, endogenous and synthetic, agonists and antagonists), and can accurately predict ligand potency based on their structure and physicochemical properties (Bonati et al 2017; Hirano et al 2015; Sovadinova et al 2006).
References
- ↑ 1.0 1.1 Okey, A. B. (2007). An aryl hydrocarbon receptor odyssey to the shores of toxicology: the Deichmann Lecture, International Congress of Toxicology-XI. Toxicol.Sci. 98, 5-38.
- ↑ Hoffman, E. C., Reyes, H., Chu, F. F., Sander, F., Conley, L. H., Brooks, B. A., and Hankinson, O. (1991). Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 252, 954-958.
- ↑ 3.0 3.1 Poland, A., Glover, E., and Kende, A. S. (1976). Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J.Biol.Chem. 251, 4936-4946.
- ↑ 4.0 4.1 Gu, Y. Z., Hogenesch, J. B., and Bradfield, C. A. (2000). The PAS superfamily: sensors of environmental and developmental signals. Annu.Rev.Pharmacol.Toxicol. 40, 519-561.
- ↑ Kewley, R. J., Whitelaw, M. L., and Chapman-Smith, A. (2004). The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int.J.Biochem.Cell Biol. 36, 189-204.
- ↑ 6.0 6.1 6.2 6.3 Fujii-Kuriyama, Y., and Kawajiri, K. (2010). Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc.Jpn.Acad.Ser.B Phys.Biol.Sci. 86, 40-53.
- ↑ 7.0 7.1 Mimura, J., and Fujii-Kuriyama, Y. (2003). Functional role of AhR in the expression of toxic effects by TCDD. Biochimica et Biophysica Acta - General Subjects 1619, 263-268.
- ↑ Giesy, J. P., Kannan, K., Blankenship, A. L., Jones, P. D., and Newsted, J. L. (2006). Toxicology of PCBs and related compounds. In Endocrine Disruption Biological Bases for Health Effects in Wildlife and Humans (D. O. Norris, and J. A. Carr, Eds.), pp. 245-331. Oxford University Press, New York.
- ↑ 9.0 9.1 9.2 Safe, S. (1994). Polychlorinated biphenyls (PCBs): Environmental impact, biochemical and toxic responses, and implications for risk assessment. Critical Reviews in Toxicology 24, 87-149.
- ↑ 10.0 10.1 10.2 Yasui, T., Kim, E. Y., Iwata, H., Franks, D. G., Karchner, S. I., Hahn, M. E., and Tanabe, S. (2007). Functional characterization and evolutionary history of two aryl hydrocarbon receptor isoforms (AhR1 and AhR2) from avian species. Toxicol.Sci. 99, 101-117.
- ↑ 11.0 11.1 Lee, J. S., Kim, E. Y., and Iwata, H. (2009). Dioxin activation of CYP1A5 promoter/enhancer regions from two avian species, common cormorant (Phalacrocorax carbo) and chicken (Gallus gallus): association with aryl hydrocarbon receptor 1 and 2 isoforms. Toxicol.Appl.Pharmacol. 234, 1-13.
- ↑ 12.0 12.1 12.2 12.3 12.4 12.5 Raucy, J. L., and Lasker, J. M. (2010). Current in vitro high throughput screening approaches to assess nuclear receptor activation. Curr. Drug Metab 11 (9), 806-814.
- ↑ 13.0 13.1 13.2 Farmahin, R., Wu, D., Crump, D., Hervé, J. C., Jones, S. P., Hahn, M. E., Karchner, S. I., Giesy, J. P., Bursian, S. J., Zwiernik, M. J., and Kennedy, S. W. (2012). Sequence and in vitro function of chicken, ring-necked pheasant, and Japanese quail AHR1 predict in vivo sensitivity to dioxins. Environ.Sci.Technol. 46, 2967-2975.
- ↑ 14.0 14.1 14.2 14.3 Farmahin, R., Manning, G. E., Crump, D., Wu, D., Mundy, L. J., Jones, S. P., Hahn, M. E., Karchner, S. I., Giesy, J. P., Bursian, S. J., Zwiernik, M. J., Fredricks, T. B., and Kennedy, S. W. (2013b). Amino acid sequence of the ligand binding domain of the aryl hydrocarbon receptor 1 (AHR1) predicts sensitivity of wild birds to effects of dioxin-like compounds. Toxicol.Sci. 131, 139-152.
- ↑ Fujisawa, N., Ikenaka, Y., Kim, E. Y., Lee, J. S., Iwata, H., and Ishizuka, M. (2012). Molecular evidence predicts aryl hydrocarbon receptor ligand insensitivity in the peregrine falcon (Falco peregrines). European Journal of Wildlife Research 58, 167-175.
- ↑ 16.0 16.1 16.2 Manning, G. E., Farmahin, R., Crump, D., Jones, S. P., Klein, J., Konstantinov, A., Potter, D., and Kennedy, S. W. (2012). A luciferase reporter gene assay and aryl hydrocarbon receptor 1 genotype predict the embryolethality of polychlorinated biphenyls in avian species. Toxicol.Appl.Pharmacol. 263, 390-399.
- ↑ Mol, T. L., Kim, E. Y., Ishibashi, H., and Iwata, H. (2012). In vitro transactivation potencies of black-footed albatross (Phoebastria nigripes) AHR1 and AHR2 by dioxins to predict CYP1A expression in the wild population. Environ.Sci.Technol. 46, 525-533.
- ↑ Yueh, M. F., Kawahara, M., and Raucy, J. (2005). Cell-based high-throughput bioassays to assess induction and inhibition of CYP1A enzymes. Toxicol. In Vitro 19 (2), 275-287.
- ↑ 19.0 19.1 Poland, A., and Knutson, J. C. (1982). 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 22, 517-554.
- ↑ 20.0 20.1 Hestermann, E. V., Stegeman, J. J., and Hahn, M. E. (2000). Relative contributions of affinity and intrinsic efficacy to aryl hydrocarbon receptor ligand potency. Toxicol. Appl. Pharmacol 168 (2), 160-172.
- ↑ 21.0 21.1 21.2 21.3 21.4 Farmahin, R., Jones, S. P., Crump, D., Hahn, M. E., Giesy, J. P., Zwiernik, M. J., Bursian, S. J., and Kennedy, S. W. (2014). Species-specific relative AHR1 binding affinities of 2,3,4,7,8-pentachlorodibenzofuran explain avian species differences in its relative potency. Comp Biochem. Physiol C. Toxicol. Pharmacol. 161C, 21-25.
- ↑ 22.0 22.1 22.2 22.3 22.4 22.5 22.6 Karchner, S. I., Franks, D. G., Kennedy, S. W., and Hahn, M. E. (2006). The molecular basis for differential dioxin sensitivity in birds: Role of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U. S. A 103 (16), 6252-6257.
- ↑ Lee, S., Shin, W. H., Hong, S., Kang, H., Jung, D., Yim, U. H., Shim, W. J., Khim, J. S., Seok, C., Giesy, J. P., and Choi, K. (2015). Measured and predicted affinities of binding and relative potencies to activate the AhR of PAHs and their alkylated analogues. Chemosphere 139, 23-29.
- ↑ Jones, S. A., Parks, D. J., and Kliewer, S. A. (2003). Cell-free ligand binding assays for nuclear receptors. Methods Enzymol. 364, 53-71.
- ↑ Gasiewicz, T. A., and Neal, R. A. (1982). The examination and quantitation of tissue cytosolic receptors for 2,3,7,8-tetrachlorodibenzo-p-dioxin using hydroxylapatite. Anal. Biochem. 124 (1), 1-11.
- ↑ Nakai, J. S., and Bunce, N. J. (1995). Characterization of the Ah receptor from human placental tissue. J Biochem. Toxicol. 10 (3), 151-159.
- ↑ Dold, K. M., and Greenlee, W. F. (1990). Filtration assay for quantitation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) specific binding to whole cells in culture. Anal. Biochem. 184 (1), 67-73.
- ↑ Perez-Romero, P., and Imperiale, M. J. (2007). Assaying protein-DNA interactions in vivo and in vitro using chromatin immunoprecipitation and electrophoretic mobility shift assays. Methods Mol. Med. 131, 123-139.
- ↑ Heid, S. E., Walker, M. K., and Swanson, H. I. (2001). Correlation of cardiotoxicity mediated by halogenated aromatic hydrocarbons to aryl hydrocarbon receptor activation. Toxicol. Sci 61 (1), 187-196.
- ↑ 30.0 30.1 Ema, M., Ohe, N., Suzuki, M., Mimura, J., Sogawa, K., Ikawa, S., and Fujii-Kuriyama, Y. (1994). Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J.Biol.Chem. 269, 27337-27343.
- ↑ 31.0 31.1 Poland, A., Palen, D., and Glover, E. (1994). Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol.Pharmacol. 46, 915-921.
- ↑ Backlund, M., and Ingelman-Sundberg, M. (2004). Different structural requirements of the ligand binding domain of the aryl hydrocarbon receptor for high- and low-affinity ligand binding and receptor activation. Mol.Pharmacol. 65, 416-425.
- ↑ Murray, I. A., Reen, R. K., Leathery, N., Ramadoss, P., Bonati, L., Gonzalez, F. J., Peters, J. M., and Perdew, G. H. (2005). Evidence that ligand binding is a key determinant of Ah receptor-mediated transcriptional activity. Arch.Biochem.Biophys. 442, 59-71.
- ↑ Pandini, A., Denison, M. S., Song, Y., Soshilov, A. A., and Bonati, L. (2007). Structural and functional characterization of the aryl hydrocarbon receptor ligand binding domain by homology modeling and mutational analysis. Biochemistry 46, 696-708.
- ↑ 35.0 35.1 35.2 Pandini, A., Soshilov, A. A., Song, Y., Zhao, J., Bonati, L., and Denison, M. S. (2009). Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry 48, 5972-5983.
- ↑ Ramadoss, P., and Perdew, G. H. (2004). Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol.Pharmacol. 66, 129-136.
- ↑ 37.0 37.1 37.2 Head, J. A., Hahn, M. E., and Kennedy, S. W. (2008). Key amino acids in the aryl hydrocarbon receptor predict dioxin sensitivity in avian species. Environ.Sci.Technol. 42, 7535-7541.
- ↑ 38.0 38.1 Denison, M. S., Soshilov, A. A., He, G., DeGroot, D. E., and Zhao, B. (2011). Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol.Sci. 124, 1-22.
- ↑ van den Berg, M., Birnbaum, L. S., Bosveld, A. T., Brunström, B., Cook, P., Feeley, M., Giesy, J. P., Hanberg, A., Hasegawa, R., Kennedy, S. W., Kubiak, T. J., Larsen, J. C., Van Leeuwen, F. X. R., Liem, A. K. D., Nolt, C., Peterson, R. E., Poellinger, L., Safe, S., Schrenk, D., Tillitt, D. E., Tysklind, M., Younes, M., Wærn, F., and Zacharewski, T. R. (1998). Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ.Health Perspect. 106, 775-792.
- ↑ Cohen-Barnhouse, A. M., Zwiernik, M. J., Link, J. E., Fitzgerald, S. D., Kennedy, S. W., Hervé, J. C., Giesy, J. P., Wiseman, S. B., Yang, Y., Jones, P. D., Wan, Y., Collins, B., Newsted, J. L., Kay, D. P., and Bursian, S. J. (2011b). Sensitivity of Japanese quail (Coturnix japonica), Common pheasant (Phasianus colchicus), and White Leghorn chicken (Gallus gallus domesticus) embryos to in ovo exposure to TCDD, PeCDF, and TCDF. Toxicol.Sci. 119, 93-103.
- ↑ Farmahin, R., Crump, D., Jones, S. P., Mundy, L. J., and Kennedy, S. W. (2013a). Cytochrome P4501A induction in primary cultures of embryonic European starling hepatocytes exposed to TCDD, PeCDF and TCDF. Ecotoxicology 22(4), 731-739.
- ↑ Hervé, J. C., Crump, D., Jones, S. P., Mundy, L. J., Giesy, J. P., Zwiernik, M. J., Bursian, S. J., Jones, P. D., Wiseman, S. B., Wan, Y., and Kennedy, S. W. (2010a). Cytochrome P4501A induction by 2,3,7,8-tetrachlorodibenzo-p-dioxin and two chlorinated dibenzofurans in primary hepatocyte cultures of three avian species. Toxicol. Sci. 113(2), 380-391.
- ↑ Hervé, J. C., Crump, D. L., McLaren, K. K., Giesy, J. P., Zwiernik, M. J., Bursian, S. J., and Kennedy, S. W. (2010b). 2,3,4,7,8-pentachlorodibenzofuran is a more potent cytochrome P4501A inducer than 2,3,7,8-tetrachlorodibenzo-p-dioxin in herring gull hepatocyte cultures. Environ. Toxicol. Chem. 29(9), 2088-2095.
- ↑ Poland, A., and Glover, E. (1973). Studies on the mechanism of toxicity of the chlorinated dibenzo-p-dioxins. Environ.Health Perspect. 5, 245-251.
- ↑ 45.0 45.1 McFarland, V. A., and Clarke, J. U. (1989). Environmental occurrence, abundance, and potential toxicity of polychlorinated biphenyl congeners: Considerations for a congener-specific analysis. Environ.Health Perspect. 81, 225-239.
- ↑ 46.0 46.1 Omiecinski, C. J., Vanden Heuvel, J. P., Perdew, G. H., and Peters, J. M. (2011). Xenobiotic metabolism, disposition, and regulation by receptors: from biochemical phenomenon to predictors of major toxicities. Toxicol.Sci. 120 Suppl 1, S49-S75.
- ↑ Swedenborg, E., and Pongratz, I. (2010). AhR and ARNT modulate ER signaling. Toxicology 268, 132-138.
- ↑ Diani-Moore, S., Ma, Y., Labitzke, E., Tao, H., David, W. J., Anderson, J., Chen, Q., Gross, S. S., and Rifkind, A. B. (2011). Discovery and biological characterization of 1-(1H-indol-3-yl)-9H-pyrido[3,4-b]indole as an aryl hydrocarbon receptor activator generated by photoactivation of tryptophan by sunlight. Chem. Biol. Interact. 193(2), 119-128.
- ↑ Wincent, E., Bengtsson, J., Mohammadi, B. A., Alsberg, T., Luecke, S., Rannug, U., and Rannug, A. (2012). Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U. S. A 109(12), 4479-4484.
- ↑ Baba, T., Mimura, J., Nakamura, N., Harada, N., Yamamoto, M., Morohashi, K., and Fujii-Kuriyama, Y. (2005). Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol.Cell Biol. 25, 10040-10051.
- ↑ Fernandez-Salguero, P. M., Pineau, T., Hilbert, D. M., McPhail, T., Lee, S. S., Kimura, S., Nebert, D. W., Rudikoff, S., Ward, J. M., and Gonzalez, F. J. (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722-726.
- ↑ Ichihara, S., Yamada, Y., Ichihara, G., Nakajima, T., Li, P., Kondo, T., Gonzalez, F. J., and Murohara, T. (2007). A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscler.Thromb.Vasc.Biol. 27, 1297-1304.
- ↑ Lahvis, G. P., Lindell, S. L., Thomas, R. S., McCuskey, R. S., Murphy, C., Glover, E., Bentz, M., Southard, J., and Bradfield, C. A. (2000). Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc.Natl.Acad.Sci U.S.A 97, 10442-10447.
- ↑ Mimura, J., Yamashita, K., Nakamura, K., Morita, M., Takagi, T. N., Nakao, K., Ema, M., Sogawa, K., Yasuda, M., Katsuki, M., and Fujii-Kuriyama, Y. (1997). Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 2, 645-654.
- ↑ Schmidt, J. V., Su, G. H., Reddy, J. K., Simon, M. C., and Bradfield, C. A. (1996). Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc.Natl.Acad.Sci U.S.A 93, 6731-6736.
- ↑ Thackaberry, E. A., Gabaldon, D. M., Walker, M. K., and Smith, S. M. (2002). Aryl hydrocarbon receptor null mice develop cardiac hypertrophy and increased hypoxia-inducible factor-1alpha in the absence of cardiac hypoxia. Cardiovasc.Toxicol. 2, 263-274.
- ↑ Zhang, N., Agbor, L. N., Scott, J. A., Zalobowski, T., Elased, K. M., Trujillo, A., Duke, M. S., Wolf, V., Walsh, M. T., Born, J. L., Felton, L. A., Wang, J., Wang, W., Kanagy, N. L., and Walker, M. K. (2010). An activated renin-angiotensin system maintains normal blood pressure in aryl hydrocarbon receptor heterozygous mice but not in null mice. Biochem.Pharmacol. 80, 197-2040.
Abnet, C.C.; Tanguay, R.L.; Heideman, W.; Peterson, R.E. 1999. Transactivation activity of human, zebrafish, and rainbow trout aryl hydrocarbon receptors expressed in COS-7 cells: Greater insight into species differences in toxic potency of polychlorinated dibenzo-p-dioxin, dibenzofuran, and biphenyl congeners. Toxicol. Appl. Pharmacol. 159, 41-51.
Andreasen, E.A.; Tanguay, R.L.; Peterson, R.E.; Heideman, W. 2002. Identification of a critical amino acid in the aryl hydrocarbon receptor. J. Biol. Chem. 277 (15), 13210-13218.
Bak, S.M.; Lida, M.; Hirano, M.; Iwata, H.; Kim, E.Y. 2013. Potencies of red seabream AHR1- and AHR2-mediated transactivation by dioxins: implications of both AHRs in dioxin toxicity. Environ. Sci. Technol. 47 (6), 2877-2885.
Clark, B.W.; Matson, C.W.; Jung, D.; Di Giulio, R.T. 2010. AHR2 mediates cardiac teratogenesis of polycyclic aromatic hydrocarbons and PCB-126 in Atlantic killifish (Fundulus heteroclitus). Aquat. Toxicol. 99, 232-240.
Doering, J.A.; Farmahin, R.; Wiseman, S.; Beitel, S.C.; Kennedy, S.W.; Giesy, J.P.; Hecker, M. 2015. Differences in activation of aryl hydrocarbon receptors of white sturgeon relative to lake sturgeon are predicted by identities of key amino acids in the ligand binding domain. Enviro. Sci. Technol. 49, 4681-4689.
Doering, J.A.; Farmahin, R.; Wiseman, S.; Kennedy, S.; Giesy J.P.; Hecker, M. 2014. Functionality of aryl hydrocarbon receptors (AhR1 and AhR2) of white sturgeon (Acipenser transmontanus) and implications for the risk assessment of dioxin-like compounds. Enviro. Sci. Technol. 48, 8219-8226.
Doering, J.A.; Giesy, J.P.; Wiseman, S.; Hecker, M. Predicting the sensitivity of fishes to dioxin-like compounds: possible role of the aryl hydrocarbon receptor (AhR) ligand binding domain. Environ. Sci. Pollut. Res. Int. 2013, 20(3), 1219-1224.
Doering, J.A.; Wiseman, S; Beitel, S.C.; Giesy, J.P.; Hecker, M. 2014. Identification and expression of aryl hydrocarbon receptors (AhR1 and AhR2) provide insight in an evolutionary context regarding sensitivity of white sturgeon (Acipenser transmontanus) to dioxin-like compounds. Aquat. Toxicol. 150, 27-35.
Duncan, D.M.; Burgess, E.A.; Duncan, I. 1998. Control of distal antennal identity and tarsal development in Drosophila by spineless-aristapedia, a homolog of the mammalian dioxin receptor. Genes Dev. 12, 1290-1303.
Eisner, B.K.; Doering, J.A.; Beitel, S.C.; Wiseman, S.; Raine, J.C.; Hecker, M. 2016. Cross-species comparison of relative potencies and relative sensitivities of fishes to dibenzo-p-dioxins, dibenzofurans, and polychlorinated biphenyls in vitro. Enviro. Toxicol. Chem. 35 (1), 173-181.
Emmons, R.B.; Duncan, D.; Estes, P.A.; Kiefel, P.; Mosher, J.T.; Sonnenfeld, M.; Ward, M.P.; Duncan, I.; Crews, S.T. 1999. The spineless-aristapedia and tango bHLH-PAS proteins interact to control antennal and tarsal development in Drosophila. Development. 126, 3937-3945.
Evans, B.R.; Karchner, S.I.; Franks, D.G.; Hahn, M.E. 2005. Duplicate aryl hydrocarbon receptor repressor genes (ahrr1 and ahrr2) in the zebrafish Danio rerio: structure, function, evolution, and AHR-dependent regulation in vivo. Arch. Biochem. Biophys. 441, 151-167.
Hahn, M.E. 2002. Aryl hydrocarbon receptors: diversity and evolution. Chemico-Biol. Interact. 141, 131-160.
Hahn, M.E.; Karchner, S.I.; Evans, B.R.; Franks, D.G.; Merson, R.R.; Lapseritis, J.M. 2006. Unexpected diversity of aryl hydrocarbon receptors in non-mammalian vertebrates: Insights from comparative genomics. J. Exp. Zool. A. Comp. Exp. Biol. 305, 693-706.
Hahn, M.E.; Poland, A.; Glover, E.; Stegeman, J.J. 1994. Photoaffinity labeling of the Ah receptor: phylogenetic survey of diverse vertebrate and invertebrate species. Arch. Biochem. Biophys. 310, 218-228.
Hansson, M.C.; Hahn, M.E. 2008. Functional properties of the four Atlantic salmon (Salmo salar) aryl hydrocarbon receptor type 2 (AHR2) isoforms. Aquat. Toxicol. 86, 121-130.
Hansson, M.C.; Wittzell, H.; Persson, K.; von Schantz, T. 2004. Unprecedented genomic diversity of AhR1 and AhR2 genes in Atlantic salmon (Salmo salar L.). Aquat. Toxicol. 68 (3), 219-232.
Karchner, S.I.; Franks, D.G.; Hahn, M.E. (2005). AHR1B, a new functional aryl hydrocarbon receptor in zebrafish: tandem arrangement of ahr1b and ahr2 genes. Biochem. J. 392 (1), 153-161.
Karchner, S.I.; Powell, W.H.; Hahn, M.E. 1999. Identification and functional characterization of two highly divergent aryl hydrocarbon receptors (AHR1 and AHR2) in the Teleost Fundulus heteroclitus. Evidence for a novel subfamily of ligand-binding basic helix loop helix-Per-ARNT-Sim (bHLH-PAS) factors. J. Biol. Chem. 274, 33814-33824.
Lahvis, G.P.; Bradfield, C.A. 1998. Ahr null alleles: distinctive or different? Biochem. Pharmacol. 56, 781-787.
Lavine, J.A.; Rowatt, A.J.; Klimova, T.; Whitington, A.J.; Dengler, E.; Beck, C.; Powell, W.H. 2005. Aryl hydrocarbon receptors in the frog Xenopus laevis: two AhR1 paralogs exhibit low affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci. 88 (1), 60-72.
Oka, K.; Kohno, S.; Ohta, Y.; Guillette, L.J.; Iguchi, T.; Katsu, Y. (2016). Molecular cloning and characterization of the aryl hydrocarbon receptors and aryl hydrocarbon receptor nuclear translocators in the American alligator. Gen. Comp. Endo. 238, 13-22.
Pongratz, I.; Mason, G.G.; Poellinger, L. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity. J. Biol. Chem. 1992, 267 (19), 13728-13734
Prasch, A.L.; Teraoka, H.; Carney, S.A.; Dong, W.; Hiraga, T.; Stegeman, J.J.; Heideman, W.; Peterson, R.E. 2003. Toxicol. Sci. Aryl hydrocarbon receptor 2 mediated 2,3,7,8-tetrachlorodibenzo-p-dioxin developmental toxicity in zebrafish. 76 (1), 138-150.
Shoots, J.; Fraccalvieri, D.; Franks, D.G.; Denison, M.S.; Hahn, M.E.; Bonati, L.; Powell, W.H. 2015. An aryl hydrocarbon receptor from the salamander Ambystoma mexicanum exhibits low sensitivity to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Enviro. Sci. Technol. 49, 6993-7001.
Tanguay, R.L.; Abnet, C.C.; Heideman, W. Peterson, R.E. (1999). Cloning and characterization of the zebrafish (Danio rerio) aryl hydrocarbon receptor1. Biochimica et Biophysica Act 1444, 35-48.
Van den Berg, M.; Birnbaum, L.; Bosveld, A.T.C.; Brunstrom, B.; Cook, P.; Feeley, M.; Giesy, J.P.; Hanberg, A.; Hasegawa, R.; Kennedy, S.W.; Kubiak, T.; Larsen, J.C.; van Leeuwen, R.X.R.; Liem, A.K.D.; Nolt, C.; Peterson, R.E.; Poellinger, L.; Safe, S.; Schrenk, D.; Tillitt, D.; Tysklind, M.; Younes, M.; Waern, F.; Zacharewski, T. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PECDFs for human and wildlife. Enviro. Hlth. Persp. 1998, 106, 775-792.
Van Tiem, L.A.; Di Giulio, R.T. 2011. AHR2 knockdown prevents PAH-mediated cardiac toxicity and XRE- and ARE-associated gene induction in zebrafish (Danio rerio). Toxicol. Appl. Pharmacol. 254 (3), 280-287.
Whitlock, J.P.; Okino, S.T.; Dong, L.Q.; Ko, H.S.P.; Clarke Katzenberg, R.; Qiang, M.; Li, W. 1996. Induction of cytochrome P4501A1: a model for analyzing mammalian gene transcription. Faseb. J. 10, 809-818.
Wirgin, I.; Roy, N.K.; Loftus, M.; Chambers, R.C.; Franks, D.G.; Hahn, M.E. 2011. Mechanistic basis of resistance to PCBs in Atlantic tomcod from the Hudson River. Science. 331, 1322-1324.
Yamauchi, M.; Kim, E.Y.; Iwata, H.; Shima, Y.; Tanabe, S. Toxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in developing red seabream (Pagrus major) embryos: an association of morphological deformities with AHR1, AHR2 and CYP1A expressions. Aquat. Toxicol. 2006, 16, 166-179.
Yasui, T.; Kim, E.Y.; Iawata, H.; Franks, D.G.; Karchner, S.I.; Hahn, M.E.; Tanabe, S. 2007. Functional characterization and evolutionary history of two aryl hydrocarbon receptor isoforms (AhR1 and AhR2) from avian species. Toxicol. Sci. 99 (1), 101-117.
Bonati, L.; Corrada, D.; Tagliabue, S.G.; Motta, S. (2017) Molecular modeling of the AhR structure and interactions can shed light on ligand-dependent activation and transformation mechanisms. Current Opinion in Toxicology 2: 42-49. https://doi.org/10.1016/j.cotox.2017.01.011.
Sovadinová, I. , Bláha, L. , Janošek, J. , Hilscherová, K. , Giesy, J. P., Jones, P. D. and Holoubek, I. (2006), Cytotoxicity and aryl hydrocarbon receptor‐mediated activity of N‐heterocyclic polycyclic aromatic hydrocarbons: Structure‐activity relationships. Environmental Toxicology and Chemistry, 25: 1291-1297. doi:10.1897/05-388R.1
Wakx A, Nedder M, Tomkiewicz-Raulet C, Dalmasso J, Chissey A, et al. 2018. Expression,
Localization, and Activity of the Aryl Hydrocarbon Receptor in the Human Placenta. Int J Mol Sci. 19(12)
Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, et al. 1994. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors.
Kall MA, Vang O, Clausen J. 1996. Effects of dietary broccoli on human in vivo drug metabolizing enzymes:
evaluation of caffeine, oestrone and chlorzoxazone metabolism. Carcinogenesis. 17(4):793–99
Miller CA. 1997. Expression of the human aryl hydrocarbon receptor complex in yeast. Activation of transcription by indole compounds. J. Biol. Chem. 272(52):32824–29
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, et al. 2011. An endogenous tumour promoting ligand of the human aryl hydrocarbon receptor. Nature. 478(7368):197–203
Adachi J, Mori Y, Matsui S, Takigami H, Fujino J, et al. 2001. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 276(34):31475–78
Marinelli L, Martin-Gallausiaux C, Bourhis J-M, B.guet-Crespel F, Blotti.re HM, Lapaque N. 2019. Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci
Rep. 9(1):643
Stobbe-Maicherski N, Wolff S, Wolff C, Abel J, Sydlik U, et al. 2013. The interleukin-6-type cytokine oncostatin M induces aryl hydrocarbon receptor expression in a STAT3-dependent manner in human HepG2 hepatoma cells. FEBS J. 280(24):6681–90
Kado S, Chang WLW, Chi AN, Wolny M, Shepherd DM, Vogel CFA. 2017. Aryl hydrocarbon receptor signaling modifies Toll-like receptor-regulated responses in human dendritic cells. Arch Toxicol. 91(5):2209–21
Bock KW. 2019. Human AHR functions in vascular tissue: Pro- and anti-inflammatory responses of AHR agonists in atherosclerosis. Biochem Pharmacol. 159:116–20
Schroeder JC, Dinatale BC, Murray IA, Flaveny CA, Liu Q, et al. 2010. The uremic toxin 3- indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 49(2):393–400
Watanabe I, Tatebe J, Namba S, Koizumi M, Yamazaki J, Morita T. 2013. Activation of aryl hydrocarbon receptor mediates indoxyl sulfate-induced monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells. Circ J. 77(1):224–30
Shackleford G, Sampathkumar NK, Hichor M, Weill L, Meffre D, et al. 2018. Involvement of Aryl hydrocarbon receptor in myelination and in human nerve sheath tumorigenesis. Proc Natl Acad Sci U S 115(6):E1319–28
Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, et al. 2008. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 118(2):640–50
Goode GD, Ballard BR, Manning HC, Freeman ML, Kang Y, Eltom SE. 2013. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB- 231 human breast cancer cell line. Int J Cancer. 133(12):2769–80
Chang JT, Chang H, Chen P-H, Lin S-L, Lin P. 2007. Requirement of aryl hydrocarbon receptor overexpression for CYP1B1 up-regulation and cell growth in human lung adenocarcinomas. Clin Cancer
Res. 13(1):38–45
Kanno Y, Takane Y, Izawa T, Nakahama T, Inouye Y. 2006. The inhibitory effect of aryl hydrocarbon receptor repressor (AhRR) on the growth of human breast cancer MCF-7 cells. Biol Pharm Bull. 29(6):1254–57
Goode G, Pratap S, Eltom SE. 2014. Depletion of the aryl hydrocarbon receptor in MDA-MB- 231 human breast cancer cells altered the expression of genes in key regulatory pathways of cancer. PLoS One. 9(6):e100103
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, et al. 2011. An endogenous tumour promoting ligand of the human aryl hydrocarbon receptor. Nature. 478(7368):197–203
Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, et al. 2016. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol Pharmacol. 90(5):674–88
Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, et al. 2014. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget. 5(4):1038–51
Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. 2010. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol Endocrinol. 24(2):359–69
Gramatzki D, Pantazis G, Schittenhelm J, Tabatabai G, K.hle C, et al. 2009. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene. 28(28):2593–2605
Subramaniam V, Ace O, Prud’homme GJ, Jothy S. 2011. Tranilast treatment decreases cell growth, migration and inhibits colony formation of human breast cancer cells. Exp Mol Pathol. 90(1):116–22
Rothhammer V, Borucki DM, Kenison JE, Hewson P, Wang Z, et al. 2018. Detection of aryl hydrocarbon receptor agonists in human samples. Sci Rep. 8(1):4970
Lin P, Chang H, Tsai W-T, Wu M-H, Liao Y-S, et al. 2003. Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol Pathol. 31(1):22–30
Barhoover MA, Hall JM, Greenlee WF, Thomas RS. 2010. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol. 77(2):195–201
Wang K, Li Y, Jiang Y-Z, Dai C-F, Patankar MS, et al. 2013. An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Lett. 340(1):63–71
Borovok N, Weiss C, Sharkia R, Reichenstein M, Wissinger B, et al. 2020. Gene and Protein Expression in Subjects With a Nystagmus-Associated AHR Mutation. Front Genet. 11:582796
List of Key Events in the AOP
Event: 149: Increase, Inflammation
Short Name: Increase, Inflammation
Key Event Component
| Process | Object | Action |
|---|---|---|
| inflammatory response | increased |
AOPs Including This Key Event
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| eukaryotic cell |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Mus musculus | Mus musculus | High | NCBI |
| Rattus norvegicus | Rattus norvegicus | High | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
Taxonomic: appears to be present broadly, with representative studies focused on mammals (humans, lab mice, lab rats).
Extensive data exists on the presence of inflammation in human (Coussens, Aggarwal, Hannhan, Mantovani..) In human, many examples of chronic inflammation leading to cancer or cancer progression exist. For instance, Helicobacter pylori infection leads to gut cancer (Wang).
Key Event Description
Inflammation is complex to define.
Villeneuve et al. (2018) analyzed the varied biological responses, provided guidance to simplify the process representing inflammation in adverse outcome pathways, and recommended 3 key steps: 1. Tissue resident cell activation 2. Increased Pro-inflammatory mediators 3. Leukocyte recruitment/activation. Tissue resident cell activation generally occurs when healthy tissue is exposed to a stressor, or when damage occurs, initiating a signal response of pro-inflammatory mediators (ex. cytokines). Pro-inflammatory mediators result in the production of lipids and proteins, signaling, and initiate leukocyte recruitment/activation. Leukocyte recruitment/activation initiate inflammation and other morphological changes.
In cancer, inflammation is a cascade of events created by the host in response to the spread of the cancer (Coussens and Werb, 2002). In response to an injury or the presence of cancer, the host heals itself through inflammation. Indeed, the activation and the migration of leukocytes (neutrophils, monocytes and eosinophils) to the wound induces the healing process. These inflammatory cells provide an extracellular matrix that forms upon which fibroblast and endothelial cells proliferate and migrate in order to recreate a normal environment. Damage to the epithelial layer initiate inflammatory reactions (Palmer et al. 2011). In cancer, this inflammatory state induces cell proliferation, increases the production of reactive oxygen species leading to oxidative DNA damage, and reduces DNA repair (Coussens and Werb, 2002). For review of inflammation caused by microplastics in mammals, see Wright and Kelly (2017).
Inflammation can be defined as the response of the organism to a tissue injury (Coussens). Indeed, in order to heal this injury, a multitude of chemical signals initiate and maintain a host response. Leukocytes (neutrophils, monocytes and eosinophils) are recruited to the site of the damage through the attraction by chemokines (TNF-α (tumour necrosis factor-α), interleukines…). A provisional extracellular matrix (ECM) is created, and fibroblast and endothelial cells proliferate and migrate to it. Wound healing is an example of physiological inflammation and is self-limiting (Coussens). In case of a dysregulation, inflammation can lead to pathologies. Inflammation can be caused by physical injury, ischemic injury, infection, exposure to toxins, or other types of trauma (Singh).
Inflammation was described as one of the hallmarks of cancer by Hannahan et al. as a response to tumor invasion through mainly two mechanisms: promoting genetic instatbility and supply pro-tumorogenic factors.
First, inflammation in cancer promotes genetic instability (Mantovani, colotta). Macrophages, in contact with the inflammatory site can be responsible of a reactive stress oxygen reaction (ROS) (Maeda, Pollard, Grivennikov). Indeed, they generate high levels of reactive oxygen and nitrogen species which produce mutagenic agents (peroxynitrite), which in turn causes DNA mutations.
Second, in inflammation, the tumor micro environment plays a critical role (Coussens). Indeed, in can supply growth factors, survival factors, proangiogenic factors, extracellular matrix-modifying enzymes that facilitate angiogenesis, invasion, and metastasis, and inductive signals that lead to activation of EMT and other hallmark-facilitating programs (Hannahan). For example, macrophages can become tumor associated macrophage which promote cell proliferation, angiogenesis, and invasion (Singh, Lin, Qian).
Moreover, chronic inflammation can also lead to tumorigenesis (Karin, Singh). Indeed, since 1863, Virchow has hypothesized that chronic inflammation causes cell proliferation (Balkwill). According to Aggarwall, several pro-inflammatory markers such as TNF and members of its superfamily, IL-1alpha, IL-1beta, IL-6, IL-8, IL-18, chemokines, MMP-9, VEGF, COX-2, and 5-LOX mediate suppression of apoptosis, proliferation, angiogenesis, invasion, and metastasis (Aggarwal).
How it is Measured or Detected
Inflammation is generally detected in histopathological examination of organs (ex. liver, intestines) or in changes in gene expression (ex. interleukins). Activation of the innate immune response and the release of various inflammatory cytokines can also be assessed (Flake and Morgan, 2017).
Several assays can be used to measure inflammation:
- Histopathology on samples. Several scoring tools exist (Goeboes)
- Measuring chemokines in the blood (ELISA, multiplex bead assays : interleukines (IL1, IL6), TNF, interferon… ) (Brenner) and histopathology samples
- Measuring Prostaglandin levels, COX-2 (ELISA
Liquid chromatography/tandem mass spectrometry, IHC) - Transcription factors : STAT3 Activation, NF-κB Activation (ELISA
RtPCR to measure mRNA - Biomarkers (white cell count, CRP) ratios, and predictive score using
- Measuring ROS(DCFDA, horseradish peroxidase (HRP)-oxidizing substrates, SOD-inhibitable reduction of cytochrome c) (Murphy).
Methods are extensively reviewed in Marchand et al and Murphy et al.
References
Flake, G.P., and Morgan, D.L. 2017. Pathology of diacetyl and 2,3-pentanedione airway lesions in a rat model of obliterative bronchiolitis. Toxicology, 388, 40–47. https://doi.org/10.1016/j.tox.2016.10.013
Palmer, S.M., Flake, G.P., Kelly, F.L., Zhang, H.L., Nugent, J.L., Kirby, P.J., Zhang, H.L., Nugent, J.L., Kirby, P.J., Foley, J.F., Gwinn, W.M., and Morgan, D.L. 2011. Severe airway epithelial injury, aberrant repair and Bronchiolitis obliterans develops after diacetyl instillation in rats. PLoS ONE, 6(3). https://doi.org/10.1371/journal.pone.0017644
Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014 Apr 10;345(2):196-202. doi: 10.1016/j.canlet.2013.08.016. Epub 2013 Aug 24. PMID: 23981572.
Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR. Tumor necrosis factor and its receptors in human ovarian cancer. Potential role in disease progression. J Clin Invest. 1993;91:2194–206.
Coussens L.M. and Werb Z. Inflammation and cancer. Nature. 2002 Dec 19-26;420(6917):860-7. doi: 10.1038/nature01322. PMID: 12490959; PMCID: PMC2803035.
Wright, S.L. and Kelly, F.J. 2017. Plastic and human health: a micro issue? Enviromental Science and Technology 51: 6634-6647.
Villeneuve, D.L., Landesmann, B., Allavena, P., Ashley, N., Bal-Price, A., Corsini, E., Halappanavar, S., Hussell, T., Laskin, D., Lawrence, T., Nikolic-Paterson, D., Pallary, M., Paini, A., Pietrs, R., Roth, R., and Tschudi-Monnet, F. 2018. Toxicological Sciences 163(2): 346-352.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010 Apr 2;141(1):39-51. doi: 10.1016/j.cell.2010.03.014. PMID: 20371344; PMCID: PMC4994190.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230.
Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6.
Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: How hot is the link? Biochem Pharmacol. 2006;72:1605–21
Singh N, Baby D, Rajguru JP, Patil PB, Thakkannavar SS, Pujari VB. Inflammation and cancer. Ann Afr Med. 2019 Jul-Sep;18(3):121-126. doi: 10.4103/aam.aam_56_18. PMID: 31417011; PMCID: PMC6704802.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44
Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability.
Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63:854–65.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8
Lin, Y., Xu, J. & Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 12, 76 (2019). https://doi.org/10.1186/s13045-019-0760-3
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010 Mar 19;140(6):883-99. doi: 10.1016/j.cell.2010.01.025. PMID: 20303878; PMCID: PMC2866629.
Murphy, M.P., Bayir, H., Belousov, V. et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab 4, 651–662 (2022). https://doi.org/10.1038/s42255-022-00591-z
Geboes K, Riddell R, Öst A, et al
A reproducible grading scale for histological assessment of inflammation in ulcerative colitis
Gut 2000;47:404-409.
Brenner DR, Scherer D, Muir K, Schildkraut J, Boffetta P, Spitz MR, Le Marchand L, Chan AT, Goode EL, Ulrich CM, Hung RJ. A review of the application of inflammatory biomarkers in epidemiologic cancer research. Cancer Epidemiol Biomarkers Prev. 2014 Sep;23(9):1729-51. doi: 10.1158/1055-9965.EPI-14-0064. Epub 2014 Jun 24. PMID: 24962838; PMCID: PMC4155060.
Event: 1262: Apoptosis
Short Name: Apoptosis
Key Event Component
| Process | Object | Action |
|---|---|---|
| apoptotic process | increased |
AOPs Including This Key Event
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| cell |
Organ term
| Organ term |
|---|
| organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Mus musculus | Mus musculus | High | NCBI |
| Rattus norvegicus | Rattus norvegicus | High | NCBI |
| Caenorhabditis elegans | Caenorhabditis elegans | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Not Otherwise Specified | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
・Apoptosis is induced in human prostate cancer cell lines (Homo sapiens) [Parajuli et al., 2014].
・Apoptosis occurs in B6C3F1 mouse (Mus musculus) [Elmore, 2007].
・Apoptosis occurs in Sprague-Dawley rat (Rattus norvegicus) [Elmore, 2007].
・Apoptosis occurs in the nematode (Caenorhabditis elegans) [Elmore, 2007].
- Apoptosis occurs in breast cancer cells, human and mouse (Parton)
Key Event Description
Apoptosis, the process of programmed cell death, is characterized by distinct morphology with DNA fragmentation and energy dependency [Elmore, 2007]. Apoptosis, also called “physiological cell death”, is involved in cell turnover, physiological involution, and atrophy of various tissues and organs [Kerr et al., 1972]. The formation of apoptotic bodies involves marked condensation of both nucleus and cytoplasm, nuclear fragmentation, and separation of protuberances [Kerr et al., 1972]. Apoptosis is characterized by DNA ladder and chromatin condensation. Several stimuli such as hypoxia, nucleotides deprivation, chemotherapeutical drugs, DNA damage, and mitotic spindle damage induce p53 activation, leading to p21 activation and cell cycle arrest [Pucci et al., 2000]. The SAHA or TSA treatment on neonatal human dermal fibroblasts (NHDFs) for 24 or 72 hrs inhibited proliferation of the NHDF cells [Glaser et al., 2003]. Considering that the acetylation of histone H4 was increased by the treatment of SAHA for 4 hrs, histone deacetylase inhibition may be involved in the inhibition of the cell proliferation [Glaser et al., 2003]. The impaired proliferation was observed in HDAC1-/- ES cells, which was rescued with the reintroduction of HDAC1 [Zupkovitz et al., 2010]. An AOP focuses existes on p21 pathway leading to apoptosis, however, alternative pathways such as NF-kappaB signaling pathways may be involved in the apoptosis of spermatocytes [Wang et al., 2017].
Apoptosis is defined as a programmed cell death. A decrease in apoptosis or a resistance to cell death is noted is described as a hallmark of cancer by Hanahan et al. It is widely admitted as an essential step in tumor proliferation (Adams, Lowe). Apoptosis occurs after activation of a number of intrinsic and extrinsic signals which activate the protease caspase system which in turn activates the destruction of the cell.
The Bcl-2 is a protein family suppressing apoptosis by binding and inhibiting two proapoptotic proteins (Bax and Bak) and transferring them to the mitochondrial outer membrane. In the absence of inhibition by Bcl2, Bax and Bak destroy the mitochondrial membrane and releases proapoptotic signaling proteins, such as cytochrome c which activated the caspase system. An increased expression of these antiapoptotic proteins (Bcl-2, Bcl-xL) occurs in cancer (Hanahan, Adams, Lowe). Several others pathways such as the loss of TP53 tumor suppressor function, or the increase of survival signals (Igf1/2), or decrease of proapoptotic factors (Bax, Bim, Puma) can also increase tumor growth (Hanahan, Juntilla).
In breast cancer a decrease in apoptosis and a resistance to cell death has been described thoroughly, especially using a dysregulation of the Bcl2 system or TP53 (Parton, Williams, Shahbandi).
How it is Measured or Detected
Apoptosis is characterized by many morphological and biochemical changes such as homogenous condensation of chromatin to one side or the periphery of the nuclei, membrane blebbing and formation of apoptotic bodies with fragmented nuclei, DNA fragmentation, enzymatic activation of pro-caspases, or phosphatidylserine translocation that can be measured using electron and cytochemical optical microscopy, proteomic and genomic methods, and spectroscopic techniques [Archana et al., 2013; Martinez et al., 2010; Taatjes et al., 2008; Yasuhara et al., 2003].
・DNA fragmentation can be quantified with comet assay using electrophoresis, where the tail length, head size, tail intensity, and head intensity of the comet are measured [Yasuhara et al., 2003].
・The apoptosis is detected with the expression alteration of procaspases 7 and 3 by Western blotting using antibodies [Parajuli et al., 2014].
・The apoptosis is measured with down-regulation of anti-apoptotic gene baculoviral inhibitor of apoptosis protein repeat containing 2 (BIRC2, or cIAP1) [Parajuli et al., 2014].
・Apoptotic nucleosomes are detected using Cell Death Detection ELISA kit, which was calculated as absorbance subtraction at 405 nm and 490 nm [Parajuli et al., 2014].
・Cleavage of PARP is detected with Western blotting [Parajuli et al., 2014].
・Caspase-3 and caspase-9 activity is measured with the enzyme-catalyzed release of p-nitroanilide (pNA) and quantified at 405 nm [Wu et al., 2016].
・Apoptosis is measured with Annexin V-FITC probes, and the relative percentage of Annexin V-FITC-positive/PI-negative cells is analyzed by flow cytometry [Wu et al., 2016].
・Apoptosis is detected with the Terminal dUTP Nick End-Labeling (TUNEL) method to assay the endonuclease cleavage products by enzymatically end-labeling the DNA strand breaks [Kressel and Groscurth, 1994].
・For the detection of apoptosis, the testes are fixed in neutral buffered formalin and embedded in paraffin. Germ cell death is visualized in testis sections by Terminal dUTP Nick End-Labeling (TUNEL) staining method [Wade et al., 2008]. The incidence of TUNEL-positive cells is expressed as the number of positive cells per tubule examined for one entire testis section per animal [Wade et al., 2008]
References
Archana, M. et al. (2013), "Various methods available for detection of apoptotic cells", Indian J Cancer 50:274-283
Elmore, S. (2007), "Apoptosis: a review of programmed cell death", Toxicol Pathol 35:495-516
Glaser, K.B. et al. (2003), "Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines", Mol Cancer Ther 2:151-163
Kerr, J.F.R. et al. (1972), "Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics", Br J Cancer 26:239-257
Kressel, M. and Groscurth, P. (1994), "Distinction of apoptotic and necrotic cell death by in situ labelling of fragmented DNA", Cell Tissue Res 278:549-556
Martinez, M.M. et al. (2010), "Detection of apoptosis: A review of conventioinal and novel techniques", Anal Methods 2:996-1004
Parajuli, K.R. et al. (2014), "Methoxyacetic acid suppresses prostate cancer cell growth by inducing growth arrest and apoptosis", Am J Clin Exp Urol 2:300-313
Pucci, B. et al. (2000), "Cell cycle and apoptosis", Neoplasia 2:291-299
Taatjes, D.J. et al. (2008), "Morphological and cytochemical determination of cell death by apoptosis", Histochem Cell Biol 129:33-43
Wade, M.G. et al. (2008), "Methoxyacetic acid-induced spermatocyte death is associated with histone hyperacetylation in rats", Biol Reprod 78:822-831
Wang, C. et al. (2017), "CD147 regulates extrinsic apoptosis in spermatocytes by modulating NFkB signaling pathways", Oncotarget 8:3132-3143
Wu, R. et al. (2016), "microRNA-497 induces apoptosis and suppressed proliferation via the Bcl-2/Bax-caspase9-caspase 3 pathway and cyclin D2 protein in HUVECs", PLoS One 11:e0167052
Yasuhara, S. et al. (2003), "Comparison of comet assay, electron microscopy, and flow cytometry for detection of apoptosis", J Histochem Cytochem 51:873-885
Zupkovitz, G. et al. (2010), "The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation", Mol Cell Biol 30:1171-1181
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230
Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007 Feb 26;26(9):1324-37. doi: 10.1038/sj.onc.1210220. PMID: 17322918; PMCID: PMC2930981.
Lowe, S., Cepero, E. & Evan, G. Intrinsic tumour suppression. Nature 432, 307–315 (2004). https://doi.org/10.1038/nature03098
Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001 Jun 23;322(7301):1528-32. doi: 10.1136/bmj.322.7301.1528. PMID: 11420276; PMCID: PMC1120573.
Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009 Nov;9(11):821-9. doi: 10.1038/nrc2728. Epub 2009 Sep 24. PMID: 19776747.
Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015 Feb 28;6(6):3519-30. doi: 10.18632/oncotarget.2792. PMID: 25784482; PMCID: PMC4414133.
Shahbandi A, Nguyen HD, Jackson JG. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer. 2020 Feb;6(2):98-110. doi: 10.1016/j.trecan.2020.01.007. Epub 2020 Feb 5. PMID: 32061310; PMCID: PMC7931175.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230
Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007 Feb 26;26(9):1324-37. doi: 10.1038/sj.onc.1210220. PMID: 17322918; PMCID: PMC2930981.
Lowe, S., Cepero, E. & Evan, G. Intrinsic tumour suppression. Nature 432, 307–315 (2004). https://doi.org/10.1038/nature03098
Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001 Jun 23;322(7301):1528-32. doi: 10.1136/bmj.322.7301.1528. PMID: 11420276; PMCID: PMC1120573.
Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009 Nov;9(11):821-9. doi: 10.1038/nrc2728. Epub 2009 Sep 24. PMID: 19776747.
Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015 Feb 28;6(6):3519-30. doi: 10.18632/oncotarget.2792. PMID: 25784482; PMCID: PMC4414133.
Shahbandi A, Nguyen HD, Jackson JG. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer. 2020 Feb;6(2):98-110. doi: 10.1016/j.trecan.2020.01.007. Epub 2020 Feb 5. PMID: 32061310; PMCID: PMC7931175.
Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001 Jun 23;322(7301):1528-32. doi: 10.1136/bmj.322.7301.1528. PMID: 11420276; PMCID: PMC1120573.
Event: 1241: Increased, Motility
Short Name: Increased, Motility
Key Event Component
| Process | Object | Action |
|---|---|---|
| cell motility | increased |
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:200 - Estrogen receptor activation leading to breast cancer | KeyEvent |
| Aop:439 - Activation of the AhR leading to metastatic breast cancer | KeyEvent |
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| eukaryotic cell |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Female | High |
| Male | High |
Cell motility has been largely described in human breast cancer cell lines, mice and fish (Stuelten)
Key Event Description
Cell motility is the capacity of cells to translocate onto a solid substratum.
In order to move several actions such as : cell–substrate adhesion, cell–cell adhesion, cell cortex rigidity (membrane and cytoskeleton), actin polymerization-mediated protrusion and actomyosin contractility (Stuelten, Lauffenburger, Montell).
Several key factors contribute to cell motility in cancer (Friedl, Lamouille, Sahail):
- Actin Cytoskeleton Dynamics: The actin cytoskeleton plays a crucial role in cell motility. Remodeling of the actin cytoskeleton is essential for cell shape changes, protrusion formation, and cell migration. This process is tightly regulated by proteins such as actin polymerization factors, focal adhesion proteins, and myosin motors.
- Cell Adhesion and Extracellular Matrix (ECM) Interactions: Integrins and other cell adhesion molecules mediate the interaction between cancer cells and the ECM. These interactions activate signaling pathways that influence cell motility. Changes in adhesion molecules can enhance or inhibit the migratory potential of breast cancer cells.
- Epithelial-Mesenchymal Transition (EMT): EMT is a biological process in which epithelial cells acquire mesenchymal characteristics, including increased motility. EMT is associated with the invasive behavior of cancer cells, allowing them to detach from the primary tumor and migrate to distant sites.
- Chemotaxis and Gradients: Cancer cells can respond to chemical gradients, a process known as chemotaxis. Growth factors and cytokines in the tumor microenvironment can attract or repel cancer cells, influencing their direction of movement.
- Proteolytic Enzymes and Matrix Metalloproteinases (MMPs): Proteolytic enzymes, especially MMPs, are involved in degrading the ECM, facilitating cancer cell invasion. The degradation of the surrounding matrix creates space for cell movement and allows cancer cells to penetrate adjacent tissues.
In breast cancer, cell motility can favor metastasis through different steps: loss of epithelial polarity, breakdown of tissue architecture, breach of the basement membrane, intravasation, extravasation, migration into new tissues, and expansion of metastatic colonies (Stuelten). For instance, an increase in invasion of the surrounding tissues and blood vessels. Once cancer cells have invaded the local tissue, they may enter the bloodstream through a process called intravasation. Subsequently, they must migrate through the vasculature to reach distant organs, a process known as extravasation (Chambers). Once in the circulation, cells utilize chemotaxis, responding to chemokines and other signals in the microenvironment, to navigate through the bloodstream and reach specific distant organs. The ability of cancer cells to home in on specific organs depends on their motility and the interactions with the target tissue (Psaila, Labelle). Once cancer cells reach a distant organ, they need to extravasate and establish micrometastases. Motility enables cancer cells to navigate through the tissue, invade the local environment, and form secondary tumor foci (Nguyen).
How it is Measured or Detected
Several assays can be used to measure cell motility, and the choice depends on the specific requirements and characteristics of the cells being studied. Here are some commonly used assays for measuring cell motility (Justus)
- Wound Healing Assay (Scratch Assay):
Principle: Create a controlled "wound" or scratch in a cell monolayer and monitor the closure of the gap over time.
Measurement: Quantify the rate of cell migration by measuring the reduction in the wound area.
- Transwell Migration Assay:
Principle: Cells migrate through a porous membrane from one side to the other in response to a chemoattractant.
Measurement: Count the number of cells that have migrated through the membrane or quantify fluorescence if cells are labeled.
- Boyden Chamber Assay:
Principle: Similar to the Transwell assay, cells migrate through a membrane towards a chemoattractant.
Measurement: Assess the migrated cells on the lower surface of the membrane.
- Time-Lapse Microscopy:
Principle: Track the movement of individual cells over time using live-cell imaging.
Measurement: Analyze cell trajectories, speed, and directionality.
- Collagen Invasion Assay:
Principle: Assess cell invasion through a three-dimensional collagen matrix.
Measurement: Quantify the extent of cell invasion into the matrix
- Fluorescence Recovery After Photobleaching (FRAP):
Principle: Measure the mobility of fluorescently labeled molecules or proteins within cells.
Measurement: Assess the recovery of fluorescence in a photobleached region over time.
- Single-Cell Tracking:
Principle: Monitor individual cell movements using time-lapse microscopy.
Measurement: Analyze parameters such as speed, persistence, and directionality for each tracked cell.
- Electric Cell-Substrate Impedance Sensing (ECIS):
Principle: Measure changes in electrical impedance as cells migrate and interact with a substrate.
Measurement: Quantify impedance-based parameters to assess cell motility.
- Bead-Based Motility Assay:
Principle: Attach beads to cells and track their movement using microscopy.
Measurement: Analyze the displacement of beads to determine cell motility.
Selecting the most appropriate assay depends on factors such as the nature of the cells, the desired readout, and the specific aspects of cell motility being investigated. Researchers often use a combination of these assays to gain a comprehensive understanding of cell motility in different contexts
References
Stuelten, C., Parent, C. & Montell, D. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer 18, 296–312 (2018). https://doi.org/10.1038/nrc.2018.15
Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014 Jun 1;(88):51046. doi: 10.3791/51046. PMID: 24962652; PMCID: PMC4186330.
Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996 Feb 9;84(3):359-69. doi: 10.1016/s0092-8674(00)81280-5. PMID: 8608589.
Chambers, A. F., Groom, A. C., & MacDonald, I. C. (2002). Dissemination and growth of cancer cells in metastatic sites. Nature Reviews Cancer, 2(8), 563–572. doi:10.1038/nrc865
Montell DJ. Morphogenetic cell movements: diversity from modular mechanical properties. Science. 2008 Dec 5;322(5907):1502-5. doi: 10.1126/science.1164073. PMID: 19056976.
Friedl, P., & Wolf, K. (2003). Tumour-cell invasion and migration: diversity and escape mechanisms. Nature Reviews Cancer, 3(5), 362–374. doi:10.1038/nrc1075
Lamouille, S., Xu, J., & Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews Molecular Cell Biology, 15(3), 178–196. doi:10.1038/nrm3758
Sahai, E. (2005). Mechanisms of cancer cell invasion. Current Opinion in Genetics & Development, 15(1), 87–96. doi:10.1016/j.gde.2004.12.002
Psaila, B., & Lyden, D. (2009). The metastatic niche: adapting the foreign soil. Nature Reviews Cancer, 9(4), 285–293. doi:10.1038/nrc2621
Labelle, M., & Hynes, R. O. (2012). The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discovery, 2(12), 1091–1099. doi:10.1158/2159-8290.CD-12-0329
Nguyen, D. X., & Bos, P. D. (2009). Massagué, J. (2009). Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer, 9(4), 274–284. doi:10.1038/nrc2622
Quail, D. F., & Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nature Medicine, 19(11), 1423–1437. doi:10.1038/nm.3394
Event: 1190: Increased, Migration (Endothelial Cells)
Short Name: Increased, Migration (Endothelial Cells)
Key Event Component
| Process | Object | Action |
|---|---|---|
| endothelial cell migration | increased |
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:200 - Estrogen receptor activation leading to breast cancer | KeyEvent |
| Aop:439 - Activation of the AhR leading to metastatic breast cancer | KeyEvent |
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| endothelial cell |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human, breast cancer cell lines
Mice
Key Event Description
Endothelial cell migration refers to the movement of endothelial cells, which are the cells lining the inner surface of blood vessels, across tissues. This dynamic process is essential for various physiological functions, including vascular development, tissue repair, and angiogenesis (Michaelis, Fonseca)..
During migration, endothelial cells undergo a series of coordinated steps, including sensing chemotactic signals, altering their cytoskeleton to form protrusions (Extension of finger-like projections (filopodia) at the leading edge of the cell to sense the environment), adhering to the extracellular matrix through molecules such as integrins,contraction (pulling the cell forward using actin) and finally detachment for movement (Michaelis, Fonseca). These movements are crucial for the remodeling and maintenance of blood vessels. This is regulated by chemical signs (VEGG, integrins) and physical cues (Michaelis, Fonseca, Norton).
The role of endothelial cell migration is in (Michaelis, Fonseca).:
- Angiogenesis: One of the primary roles of endothelial cell migration is in angiogenesis, the formation of new blood vessels. In response to signals from growth factors like vascular endothelial growth factor (VEGF), endothelial cells migrate towards the site of angiogenesis, contributing to the expansion of the vascular network (Michaelis, Lamalis).
- Tissue Repair: Endothelial cell migration is crucial for repairing damaged blood vessels. In response to injury or inflammation, endothelial cells migrate to the site of damage, facilitating the restoration of vascular integrity (Michaelis).
- Vascular Development: During embryonic development, endothelial cell migration is essential for the formation and remodeling of blood vessels (Scarpa). This process helps establish the intricate vascular network required for organ development.
- Immune Response: Endothelial cells play a role in immune responses by facilitating the migration of immune cells across blood vessel walls to sites of infection or injury (Sturtzel).
- Lymphangiogenesis: Endothelial cell migration is involved in lymphangiogenesis, the formation of new lymphatic vessels. This process is crucial for fluid drainage, immune surveillance, and can also play a role in cancer metastasis (Pengchung).
- Wound Healing:Endothelial cells contribute to wound healing by migrating to the site of injury and participating in the formation of new blood vessels, a process known as neovascularization (Lamalis, Amersfoort).
- Cancer Metastasis: In cancer, endothelial cell migration is hijacked by tumors to support their growth and metastasis. Tumor cells release angiogenic factors, inducing the migration of endothelial cells to form new blood vessels that supply nutrients to the growing tumor (Lamalis).
How it is Measured or Detected
Assays used to study endothelial cell migration (Guo):
- Boyden chamber: evaluates the ability of cells to migrate through a porous membrane towards a chemoattractant (substance that attracts cells) placed in the lower chamber.
- Scratch wound assay: collective movement of endothelial cells to close a "wound" created by scratching a confluent monolayer of cells.
- Microfluidic assay: microfluidic channels to create controlled environments that mimic the physiological flow conditions experienced by endothelial cells in vivo (Shih)
- Tube formation: assays evaluate the ability of endothelial cells to form tube-like structures, mimicking the process of blood vessel formation (angiogenesis) (Guo)
- Collagen Invasion Assay: Assess the invasive capacity of endothelial cells through a three-dimensional collagen matrix
- Time-lapse microscopy: using live-cell imaging
- 3D spheroid migration
- In vivo: vessel density in fat pads
References
Mierke CT. Role of the endothelium during tumor cell metastasis: is the endothelium a barrier or a promoter for cell invasion and metastasis? J Biophys. 2008;2008:183516. doi: 10.1155/2008/183516. Epub 2009 Mar 5. PMID: 20107573; PMCID: PMC2809021.
Michaelis UR. Mechanisms of endothelial cell migration. Cell Mol Life Sci. 2014 Nov;71(21):4131-48. doi: 10.1007/s00018-014-1678-0. Epub 2014 Jul 20. PMID: 25038776.
Guo S, Lok J, Liu Y, Hayakawa K, Leung W, Xing C, Ji X, Lo EH. Assays to examine endothelial cell migration, tube formation, and gene expression profiles. Methods Mol Biol. 2014;1135:393-402. doi: 10.1007/978-1-4939-0320-7_32. PMID: 24510881; PMCID: PMC4035906.
Shih, HC., Lee, TA., Wu, HM. et al. Microfluidic Collective Cell Migration Assay for Study of Endothelial Cell Proliferation and Migration under Combinations of Oxygen Gradients, Tensions, and Drug Treatments. Sci Rep 9, 8234 (2019). https://doi.org/10.1038/s41598-019-44594-5
Fonseca CG, Barbacena P, Franco CA. Endothelial cells on the move: dynamics in vascular morphogenesis and disease. Vasc Biol. 2020 Jul 2;2(1):H29-H43. doi: 10.1530/VB-20-0007. PMID: 32935077; PMCID: PMC7487603.
Yu P, Wu G, Lee HW, Simons M. Endothelial Metabolic Control of Lymphangiogenesis. Bioessays. 2018 Jun;40(6):e1700245. doi: 10.1002/bies.201700245. Epub 2018 May 11. PMID: 29750374; PMCID: PMC6237195.
Amersfoort, J., Eelen, G. & Carmeliet, P. Immunomodulation by endothelial cells — partnering up with the immune system?. Nat Rev Immunol 22, 576–588 (2022). https://doi.org/10.1038/s41577-022-00694-4
Sturtzel C. Endothelial Cells. Adv Exp Med Biol. 2017;1003:71-91. doi: 10.1007/978-3-319-57613-8_4. PMID: 28667554.
Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007 Mar 30;100(6):782-94. doi: 10.1161/01.RES.0000259593.07661.1e. PMID: 17395884.
Scarpa E, Mayor R. Collective cell migration in development. J Cell Biol. 2016 Jan 18;212(2):143-55. doi: 10.1083/jcb.201508047. PMID: 26783298; PMCID: PMC4738384.
Michaelis UR. Mechanisms of endothelial cell migration. Cell Mol Life Sci. 2014 Nov;71(21):4131-48. doi: 10.1007/s00018-014-1678-0. Epub 2014 Jul 20. PMID: 25038776.
Norton KA, Popel AS. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci Rep. 2016 Nov 14;6:36992. doi: 10.1038/srep36992. PMID: 27841344; PMCID: PMC5107954.
Event: 1196: Increased, Invasion
Short Name: Increased, Invasion
Key Event Component
| Process | Object | Action |
|---|---|---|
| epithelial cell | increased |
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:200 - Estrogen receptor activation leading to breast cancer | KeyEvent |
| Aop:439 - Activation of the AhR leading to metastatic breast cancer | KeyEvent |
| Aop:495 - Androgen receptor activation leading to prostate cancer | KeyEvent |
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Mice
Key Event Description
Cell invasion refers to the active movement of cells into and through tissues, barriers, or extracellular matrices (ECM) (Friedl). It involves a series of coordinated processes by which cells penetrate physical barriers, navigate through the extracellular environment, and potentially reach distant locations (Hynes). It is regulated by growth factors (VEGF), signaling pathways and cell-cell interactions.
Key Steps in Cell Invasion:
- Detachment: Detachment of cells to the extracellular matrix (ECM) or neighboring cells through interactions with adhesion molecules, including integrins and cadherins.
- Proteolysis: Degradation of ECM components by proteolytic enzymes, such as matrix metalloproteinases (MMPs), secreted by invasive cells. This process creates pathways for cell movement.
- Motility: Dynamic changes in the cell's cytoskeleton, involving the formation of actin-rich structures like lamellipodia and filopodia, which facilitate cell movement.
- Intravasation: Invasion of cells into blood vessels or lymphatic vessels, allowing them to enter the circulatory system and potentially spread to distant sites.
- Extravasation: Exit of invasive cells from the bloodstream or lymphatic vessels at a secondary site, facilitating colonization and the formation of secondary tumors.
- Adhesion: Cells form new attachments to the ECM at the leading edge, allowing for continued movement.
There are many roles for cell invasion:
- Development and Tissue Repair: Cell invasion is crucial during embryonic development for processes such as tissue patterning and organ formation. In adults, invasion is essential for tissue repair and regeneration.
- Embryonic development: During development, cells migrate to form different organs and tissues, shaping the intricate structure of the organism (Heisenberg).
- Immune Response: Immune cells use invasion to migrate to sites of infection or injury, where they participate in immune responses.
- Angiogenesis: Endothelial cells migrate to form new blood vessels, delivering oxygen and nutrients to growing tissues or healing wounds (Carmeliet, Lamalice).
- Wound Healing: Invasive migration of cells is essential for wound healing, allowing cells to move into the wounded area and contribute to tissue repair (Grinnell).
- Cancer Metastasis: In cancer, invasion is a hallmark of malignancy and a critical step in metastasis. Cancer cells acquire the ability to invade surrounding tissues, enter blood or lymphatic vessels, and establish secondary tumors at distant sites (Krakhmal).
How it is Measured or Detected
Several assays can be used to study cell invasion (Justus):
- Transwell Invasion Assay: Cells migrate through a porous membrane coated with ECM proteins toward a chemoattractant (Hulkower).
- Boyden Chamber Assay: cell migration and invasion through a porous membrane in response to a gradient of chemoattractants.
- 3D Spheroid Invasion Assay: spheroids embedded in a 3D matrix, and invasion is assessed as cells migrate out from the spheroid into the surrounding matrix (Pijuan).
- Collagen Invasion Assay: Cells invade through a collagen matrix, simulating the extracellular environment.
- Matrigel Invasion Assay: Cells invade through Matrigel, a basement membrane matrix rich in ECM proteins.
- Zymography: Assess the activity of matrix metalloproteinases (MMPs), enzymes involved in ECM degradation and cell invasion.
- Electric Cell-Substrate Impedance Sensing (ECIS): Measure changes in electrical impedance as cells invade and interact with a substrate.
- Microfluidic Invasion Assays: Use microfluidic devices to create controlled environments for studying cell invasion (Fonseca).
- In Vivo Invasion Assays: Intravital imaging or xenograft models to study cell invasion in vivo.
References
Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014 Jun 1;(88):51046. doi: 10.3791/51046. PMID: 24962652; PMCID: PMC4186330.
Fonseca CG, Barbacena P, Franco CA. Endothelial cells on the move: dynamics in vascular morphogenesis and disease. Vasc Biol. 2020 Jul 2;2(1):H29-H43. doi: 10.1530/VB-20-0007. PMID: 32935077; PMCID: PMC7487603.
Pijuan J, Barceló C, Moreno DF, Maiques O, Sisó P, Marti RM, Macià A, Panosa A. In vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front Cell Dev Biol. 2019 Jun 14;7:107. doi: 10.3389/fcell.2019.00107. PMID: 31259172; PMCID: PMC6587234.
Hulkower KI, Herber RL. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics. 2011 Mar 11;3(1):107-24. doi: 10.3390/pharmaceutics3010107. PMID: 24310428; PMCID: PMC3857040.
Friedl, P., & Weigelin, B. (2008). Interstitial cell migration and invasion in tumorous environments: Past, present and future. Cell adhesion & migration, 2(1), 115-125. https://pathsocjournals.onlinelibrary.wiley.com/doi/full/10.1002/path.3031
Hynes, R. O. (2009). The extracellular matrix in action. Cell, 137(5), 910-921. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4185430/
Krakhmal NV, Zavyalova MV, Denisov EV, Vtorushin SV, Perelmuter VM. Cancer Invasion: Patterns and Mechanisms. Acta Naturae. 2015 Apr-Jun;7(2):17-28. PMID: 26085941; PMCID: PMC4463409.
Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007 Mar 30;100(6):782-94. doi: 10.1161/01.RES.0000259593.07661.1e. PMID: 17395884.
Heisenberg, C. P., & Bellairs, R. (2013). Cell migration in development and disease. Nature reviews. Molecular cell biology, 14(7), 481-494. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4457291/
Grinnell, F. (2003). Fibroblast biology: From contraction to proliferation. Journal of cell physiology, 197(1), 301-303. https://pubmed.ncbi.nlm.nih.gov/8106541/
Carmeliet, P., & Jain, R. K. (2011). Angiogenesis in disease and the angiogenic switch. Nature medicine, 17(7), 755-763
Event: 1376: Increase, angiogenesis
Short Name: Increase, angiogenesis
Key Event Component
| Process | Object | Action |
|---|---|---|
| Breast carcinoma | VEGF-A complex | increased |
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:217 - Gastric ulcer formation | KeyEvent |
| Aop:439 - Activation of the AhR leading to metastatic breast cancer | KeyEvent |
Biological Context
| Level of Biological Organization |
|---|
| Tissue |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Male | High |
| Female | High |
Human
Mice
Key Event Description
Angiogenesis is the physiological process through which new blood vessels form from existing vessels. This complex and tightly regulated process involves the proliferation and migration of endothelial cells, the remodeling of the extracellular matrix, and the recruitment of pericytes and smooth muscle cells.
Key Steps in Angiogenesis:
- Stimulus for Angiogenesis: Angiogenesis is triggered by specific signals, such as growth factors, released in response to tissue hypoxia, injury, or other physiological needs.
- Activation of Endothelial Cells: Endothelial cells in existing blood vessels become activated in response to angiogenic signals, leading to changes in gene expression and cell behavior.
- Proliferation and Migration: Activated endothelial cells proliferate and migrate toward the angiogenic stimulus, guided by chemotactic signals.
- Tube Formation: Endothelial cells organize into tube-like structures, forming capillaries. This process involves the creation of lumens within the tubes.
- Vessel Maturation: The newly formed vessels undergo maturation processes, including the recruitment of pericytes and smooth muscle cells. This maturation is crucial for the stability and functionality of the vasculature.
- Integration with Circulatory System: The newly formed blood vessels integrate into the existing circulatory system, providing increased blood flow to the target tissues.
Angiogenesis is regulated by both pro and anti-angiogenic factors. The most common pro angiogenic factors are VEGF and FGF (Folkman).
Angiogenesis is a fundamental mechanism in development, tissue repair, and various pathological conditions, including cancer :
- Development: During embryonic development, angiogenesis is critical for establishing the vascular network necessary for organ and tissue formation (Ribatti).
- Tissue Repair and Regeneration: Angiogenesis plays a key role in tissue repair and regeneration after injury or damage. The formation of new blood vessels helps supply nutrients and oxygen to the healing tissue (Ribatti).
- Menstrual Cycle and Pregnancy: In the female reproductive system, angiogenesis is a normal part of the menstrual cycle and is essential for the development of the placenta during pregnancy (Hoier).
- Inflammatory Response: Angiogenesis is involved in the inflammatory response, facilitating the influx of immune cells to sites of infection or injury.
- Cancer Growth and Metastasis : In cancer, angiogenesis is hijacked by tumors to support their growth and metastasis. Tumors release pro-angiogenic factors, promoting the formation of new blood vessels that supply nutrients and oxygen to the growing cancer cells (Nishida).
- Ischemic Diseases: Angiogenesis is a therapeutic target in diseases involving inadequate blood supply, such as ischemic heart disease and peripheral artery disease (Ferrara)
How it is Measured or Detected
Several assays are commonly employed (Staton, Stryker, Irvin):
- Endothelial Cell Proliferation Assays: Assays like BrdU (bromodeoxyuridine) incorporation, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, or EdU (5-ethynyl-2'-deoxyuridine) incorporation can be used
- Endothelial Cell Migration Assays: Transwell migration assays, scratch/wound healing assays, or microfluidic devices to study directed migration.
- Tube Formation Assay: Assess the ability of endothelial cells to form capillary-like structures (Stryker)
- Chorioallantoic Membrane (CAM) Assay: In vivo assay utilizing the chick embryo CAM to observe angiogenesis (Staton).
- Matrigel Plug Assay: In vivo assay involving the subcutaneous injection of Matrigel containing angiogenic inducers or cells (Tahergorabi).
- Aortic Ring Assay
- Corneal Neovascularization Assay
- Angiogenesis Imaging: as confocal microscopy or intravital microscopy to visualize blood vessel formation.
- Quantitative PCR (qPCR) for Angiogenic Markers
- ELISA for Angiogenic Factors
References
Staton CA, Stribbling SM, Tazzyman S, Hughes R, Brown NJ, Lewis CE. Current methods for assaying angiogenesis in vitro and in vivo. Int J Exp Pathol. 2004 Oct;85(5):233-48. doi: 10.1111/j.0959-9673.2004.00396.x. PMID: 15379956; PMCID: PMC2517524.
Irvin MW, Zijlstra A, Wikswo JP, Pozzi A. Techniques and assays for the study of angiogenesis. Exp Biol Med (Maywood). 2014 Nov;239(11):1476-88. doi: 10.1177/1535370214529386. Epub 2014 May 28. PMID: 24872440; PMCID: PMC4216737.
Tahergorabi Z, Khazaei M. A review on angiogenesis and its assays. Iran J Basic Med Sci. 2012 Nov;15(6):1110-26. PMID: 23653839; PMCID: PMC3646220.
Stryker ZI, Rajabi M, Davis PJ, Mousa SA. Evaluation of Angiogenesis Assays. Biomedicines. 2019 May 16;7(2):37. doi: 10.3390/biomedicines7020037. PMID: 31100863; PMCID: PMC6631830.
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213-9. doi: 10.2147/vhrm.2006.2.3.213. PMID: 17326328; PMCID: PMC1993983.
Ribatti, C. (2016). The crucial role of angiogenesis in embryogenesis. Life Sciences, 157, 17-22
Ribatti, C. (2014). The role of angiogenesis in wound healing. Journal of Vascular Research, 51(1), 2-11.
Reynolds, L. P., & Grazul-Balsas, J. L. (2010). Angiogenesis in the corpus luteum. Endocrine Reviews, 31(2), 226-240. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2820779/
Hoier, J. D., & Hellström, M. (2014). Regulation of skeletal muscle angiogenesis by exercise training. Journal of Physiology, 592(11), 2523-2533. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4104895/
Ferrara, N. (2005). VEGF as a therapeutic target in cancer. Cancer Cell, 8(6), 399-407.
Folkman, J. (2002). Angiogenesis: an essential step in tumor progression. Seminars in Oncology, 29(6), 315-322. [https://pubmed
Staton CA, Stribbling SM, Tazzyman S, Hughes R, Brown NJ, Lewis CE. Current methods for assaying angiogenesis in vitro and in vivo. Int J Exp Pathol. 2004 Oct;85(5):233-48. doi: 10.1111/j.0959-9673.2004.00396.x. PMID: 15379956; PMCID: PMC2517524.
Irvin MW, Zijlstra A, Wikswo JP, Pozzi A. Techniques and assays for the study of angiogenesis. Exp Biol Med (Maywood). 2014 Nov;239(11):1476-88. doi: 10.1177/1535370214529386. Epub 2014 May 28. PMID: 24872440; PMCID: PMC4216737.
Tahergorabi Z, Khazaei M. A review on angiogenesis and its assays. Iran J Basic Med Sci. 2012 Nov;15(6):1110-26. PMID: 23653839; PMCID: PMC3646220.
Stryker ZI, Rajabi M, Davis PJ, Mousa SA. Evaluation of Angiogenesis Assays. Biomedicines. 2019 May 16;7(2):37. doi: 10.3390/biomedicines7020037. PMID: 31100863; PMCID: PMC6631830.
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213-9. doi: 10.2147/vhrm.2006.2.3.213. PMID: 17326328; PMCID: PMC1993983.
Ribatti, C. (2016). The crucial role of angiogenesis in embryogenesis. Life Sciences, 157, 17-22
Ribatti, C. (2014). The role of angiogenesis in wound healing. Journal of Vascular Research, 51(1), 2-11.
Reynolds, L. P., & Grazul-Balsas, J. L. (2010). Angiogenesis in the corpus luteum. Endocrine Reviews, 31(2), 226-240. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2820779/
Hoier, J. D., & Hellström, M. (2014). Regulation of skeletal muscle angiogenesis by exercise training. Journal of Physiology, 592(11), 2523-2533. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4104895/
Ferrara, N. (2005). VEGF as a therapeutic target in cancer. Cancer Cell, 8(6), 399-407.
Folkman, J. (2002). Angiogenesis: an essential step in tumor progression. Seminars in Oncology, 29(6), 315-322. [https://pubmed
Event: 1971: Increased, tumor growth
Short Name: tumor growth
Key Event Component
| Process | Object | Action |
|---|---|---|
| Breast carcinoma | BRCA1-A complex | increased |
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:439 - Activation of the AhR leading to metastatic breast cancer | KeyEvent |
Biological Context
| Level of Biological Organization |
|---|
| Organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human, mice
Key Event Description
Tumor growth refers to the increase in size of a cancer due to the uncontrolled proliferation of cells. The mechanisms have been detailed in Hanahan et al. hallmarks of cancer:
- Initiation: Tumor growth often begins with the initiation of genetic alterations in normal cells. This can result from mutations caused by various factors such as exposure to carcinogens, genetic predisposition, or viral infections.
- Uncontrolled Cell Proliferation: One of the hallmark features of tumor growth is uncontrolled cell division. Initiating mutations in key regulatory genes, such as oncogenes and tumor suppressor genes, disrupt normal cell cycle control, leading to continuous and unregulated cell proliferation. The PI3K/AKT/mTOR pathway regulates cell growth, proliferation, and survival. Mutations in genes like PTEN, a negative regulator of this pathway, can lead to its hyperactivation, promoting tumor growth (Janaku, Paplomatta). The MAPK is involved in cell proliferation, differentiation, and survival. Mutations in genes like BRAF and KRAS can activate this pathway, contributing to uncontrolled cell growth and tumor development (Steelman, Guo).
- Angiogenesis: Tumors require a blood supply for sustained growth. Angiogenesis, the formation of new blood vessels, is induced by the tumor to ensure a nutrient and oxygen supply. Tumor cells release pro-angiogenic factors, promoting the development of a network of blood vessels within and around the tumor (Nishida).
- Metabolic Adaptations: Tumor cells often exhibit altered metabolism, characterized by increased glycolysis even in the presence of oxygen (Warburg effect). This metabolic shift supports the high energy demands of rapidly dividing cells (Pham).
- Tumor Microenvironment: Tumor growth involves interactions with the surrounding microenvironment, including stromal cells, immune cells, and the extracellular matrix. Tumor cells can influence their microenvironment to promote their survival and expansion. Fibroblasts transform into cancer associated fibroblasts to support tumor growth by producing growth factors and promoting angiogenesis (Asif).
- Immune Evasion: Malignant tumors can develop mechanisms to evade the immune system. This may involve downregulation of antigens, inhibitory signals to immune cells, or the recruitment of immunosuppressive cells, allowing the tumor to escape immune detection and attack (Hiam).
- Invasion and Metastasis: Malignant tumors can invade nearby tissues and, in advanced stages, metastasize to distant organs. Invasion involves the penetration of tumor cells into surrounding tissues, while metastasis is the spread of cancer cells to other parts of the body via the bloodstream or lymphatic system.
- Tumor Dormancy: In some cases, tumor growth may enter a state of dormancy, where the proliferation of cancer cells is temporarily halted. Dormant tumors can later resume growth, posing challenges in terms of early detection and treatment (Endo).
Detailed here are key molecular mechanisms associated with breast tumor growth (Hanahan):
- Genetic Mutations: Genetic alterations in key oncogenes (e.g., HER2, MYC, PIK3CA) promote cell proliferation whereas mutations in tumor suppressor genes (e.g., TP53, BRCA1, BRCA2) remove inhibitory controls on cell growth. (Knudson)
- Hormone Receptor Signaling: ER-positive breast cancers (70% of cancers) respond to estrogen stimulation, promoting cell proliferation. Endocrine therapies targeting ER signaling are effective in treating these cancers (Elikatkin).
- HER2/Neu overexpression : Amplification or overexpression of the human epidermal growth factor receptor 2 (HER2) promotes cell growth and survival (Slamon, Elikatkin).
- PI3K/AKT/mTOR Pathway Activation: Mutations in the PIK3CA gene or activation of PI3K signaling pathway promotes cell survival and proliferation. Phosphoinositide 3-kinase (PI3K) activation leads to downstream signaling through AKT and mTOR, promoting cell growth and protein synthesis (Janku, Paplomata)
- MAPK pathway: This pathway is involved in cell proliferation, differentiation, and survival. Mutations in this pathway can also contribute to breast cancer development (Steelman).
- Cell Cycle Regulation: Dysregulation of cyclin-dependent kinase (CDK) and cyclin complexes controls the cell cycle progression. Inactivation of the p16 tumor suppressor and retinoblastoma protein (pRB) pathway contributes to uncontrolled cell cycle progression (Witkiewicz).
- Apoptosis Evasion: Overexpression of anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL) inhibits programmed cell death. Mutations or inactivation of pro-apoptotic proteins (e.g., p53) hinders apoptotic responses.
- Angiogenesis Stimulation: Vascular endothelial growth factor (VEGF) and its receptors stimulate angiogenesis, ensuring a blood supply for tumor growth. Hypoxia-inducible factor 1-alpha (HIF-1α) activates angiogenic responses in low-oxygen conditions.
- Epithelial-Mesenchymal Transition (EMT): Downregulation of adhesion molecules (e.g., E-cadherin) leads to increased cell mobility. Acquisition of mesenchymal characteristics enhances the ability of tumor cells to invade surrounding tissues (Drasin).
- Extracellular Matrix (ECM) Remodeling: Overexpression of MMPs facilitates ECM degradation, enabling tumor invasion.
- Metastasis Formation: Tumor cells invade surrounding tissues and enter blood or lymphatic vessels. Ability of tumor cells to survive in the bloodstream. Tumor cells exit circulation, invade distant tissues, and establish secondary tumors.
How it is Measured or Detected
Many different assays can be used to measure tumor growth directly:
- Clinical measurement and palpation
- Histopathology with fluorescence imaging, dyes or weight
- Serum Biomarkers
- Imagery using caliper measurement on Magnetic Resonance Imaging (MRI), Computed Tomography (CT), Positron Emission Tomography (PET), or ultrasound can provide detailed images for volume calculation.
- Positron Emission Tomography (PET) Imaging : measurement of metabolic activity using radioactive tracers.
- In vivo models: xenograft tumor models, orthotopic models, genetically engineered mouse models
Indirect assays can also be used:
- Bioluminescence Imaging (BLI): Measurement of light emitted by luciferase-expressing tumor cells.
- Flow Cytometry: Quantification of tumor cells based on DNA content.
- Cell Proliferation Assays (MTT/MTS, BrdU)
- Colony formation
References
Asif PJ, Longobardi C, Hahne M, Medema JP. The Role of Cancer-Associated Fibroblasts in Cancer Invasion and Metastasis. Cancers (Basel). 2021 Sep 21;13(18):4720. doi: 10.3390/cancers13184720. PMID: 34572947; PMCID: PMC8472587.
Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014 May 7;16(3):207. doi: 10.1186/bcr3652. PMID: 25223380; PMCID: PMC4076637.
Eliyatkın N, Yalçın E, Zengel B, Aktaş S, Vardar E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J Breast Health. 2015 Apr 1;11(2):59-66. doi: 10.5152/tjbh.2015.1669. PMID: 28331693; PMCID: PMC5351488.
Phan LM, Yeung SC, Lee MH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014 Mar;11(1):1-19. doi: 10.7497/j.issn.2095-3941.2014.01.001. PMID: 24738035; PMCID: PMC3969803.
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213-9. doi: 10.2147/vhrm.2006.2.3.213. PMID: 17326328; PMCID: PMC1993983.
Drasin, D.J., Robin, T.P. & Ford, H.L. Breast cancer epithelial-to-mesenchymal transition: examining the functional consequences of plasticity. Breast Cancer Res 13, 226 (2011). https://doi.org/10.1186/bcr3037
Paplomata E, O'Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014 Jul;6(4):154-66. doi: 10.1177/1758834014530023. PMID: 25057302; PMCID: PMC4107712.
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y and Hu LL: ERK/MAPK signalling pathway and tumorigenesis (Review). Exp Ther Med 19: 1997-2007, 2020
Hiam-Galvez, K.J., Allen, B.M. & Spitzer, M.H. Systemic immunity in cancer. Nat Rev Cancer 21, 345–359 (2021). https://doi.org/10.1038/s41568-021-00347-z
Endo H, Inoue M. Dormancy in cancer. Cancer Sci. 2019 Feb;110(2):474-480. doi: 10.1111/cas.13917. Epub 2019 Jan 11. PMID: 30575231; PMCID: PMC6361606.
Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J., Wong, S. G., Keith, D. E., ... & McGuire, W. L. (1989). Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science (New York, N.Y.), 248(4960), 787-792. https://pubmed.ncbi.nlm.nih.gov/2470152/
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57-70. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446472/
Knudson, A. G. (2000). Two-hit hypothesis for inherited breast cancer: an update. Carcinogenesis, 21(3), 439-448. https://pubmed.ncbi.nlm.nih.gov/9212799/
Janku, F., Yap, T. A., & Westin, J. (2018). Targeting the PI3K pathway in human cancer: rationale and emerging clinical landscapes. Journal of Clinical Oncology, 36(15), 1550-1562. https://pubmed.ncbi.nlm.nih.gov/29508857/
Steelman, L. S., Chappell, W. P., deCarvalho, T. B., Lowe, S., & Davies, M. (2004. Ras/Raf/MEK/
List of Adverse Outcomes in this AOP
Event: 1982: metastatic breast cancer
Short Name: Metastasis, Breast Cancer
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:443 - DNA damage and mutations leading to Metastatic Breast Cancer | AdverseOutcome |
| Aop:439 - Activation of the AhR leading to metastatic breast cancer | AdverseOutcome |
Stressors
| Name |
|---|
| Ethyl alcohol |
Biological Context
| Level of Biological Organization |
|---|
| Organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human and other cells in culture | human and other cells in culture | High | NCBI |
| human | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Increased metastasis of cancerous cells is known to be highly conserved throughout evolution and is present from humans to invertebrates.
Key Event Description
Processs: metastasis of cancer cells Object:metastasis Process:Increased
Biological state:
Metastasis, the process by which cancer cells spread from their site of origin to distant organs or tissues, is a complex and multifaceted biological phenomenon that poses a significant challenge in cancer management. Cancer metastasis represents a critical stage in the progression of the disease, often leading to poorer patient outcomes and decreased survival rates. Understanding the molecular and cellular mechanisms underlying metastasis is crucial for developing effective therapeutic strategies to combat advanced-stage cancers.
At the biological level, metastasis involves a series of sequential steps that cancer cells must undergo to successfully disseminate and colonize distant sites within the body. These steps include local invasion of surrounding tissues by cancer cells, intravasation into nearby blood or lymphatic vessels, survival and transport through the circulation, extravasation into distant tissues, and establishment of secondary tumors through proliferation and angiogenesis. Each of these steps is regulated by a complex interplay of genetic, epigenetic, and microenvironmental factors that influence the invasive and migratory properties of cancer cells.
The metastatic process is driven by a variety of molecular alterations that confer cancer cells with the ability to invade and metastasize. Key molecular mechanisms implicated in metastasis include dysregulated signaling pathways involved in cell adhesion, motility, and invasion, as well as genetic mutations and epigenetic modifications that promote tumor progression and metastatic spread. For example, alterations in genes encoding cell adhesion molecules such as E-cadherin, integrins, and cadherins can disrupt cell-cell and cell-matrix interactions, facilitating the detachment and dissemination of cancer cells from the primary tumor site.
Furthermore, the tumor microenvironment plays a critical role in regulating the metastatic behavior of cancer cells. Stromal cells, immune cells, and extracellular matrix components within the tumor microenvironment interact dynamically with cancer cells to modulate their invasive and migratory properties. Additionally, factors such as hypoxia, inflammation, and angiogenesis contribute to the formation of a pro-metastatic niche that supports the survival and outgrowth of disseminated cancer cells at distant sites.
In summary, metastasis is a complex biological process driven by genetic, molecular, and microenvironmental factors that enable cancer cells to spread and establish secondary tumors in distant organs or tissues. Understanding the underlying mechanisms of metastasis is essential for the development of targeted therapies aimed at disrupting key molecular pathways involved in this process, ultimately improving outcomes for patients with advanced-stage cancers.
Biological compartment
Organs,Cellular
Role in general biology
Metastasis, although primarily studied in the context of cancer biology, also has relevance in general biology as it reflects fundamental biological processes such as cell migration, invasion, and tissue remodeling. Understanding these processes not only sheds light on cancer progression but also provides insights into various physiological and pathological phenomena in multicellular organisms.
1. Cell Migration: Cell migration is a fundamental process in various biological contexts, including embryonic development, wound healing, and immune responses. Metastasis involves the migration of cancer cells from the primary tumor to distant sites within the body, exploiting mechanisms similar to those used by normal cells during migration. Studying cancer metastasis can provide valuable insights into the molecular mechanisms underlying cell migration, including changes in cytoskeletal dynamics, cell adhesion, and signaling pathways that regulate cell motility.
2. Invasion and Extravasation: Cancer metastasis requires cancer cells to invade surrounding tissues, intravasate into blood or lymphatic vessels, survive in the circulation, and extravasate into distant tissues. These processes involve complex interactions between cancer cells and the surrounding microenvironment, including extracellular matrix components, immune cells, and stromal cells. Understanding the mechanisms of invasion and extravasation in cancer metastasis can provide insights into how cells navigate and interact with their microenvironment under physiological and pathological conditions.
3. Tissue Remodeling and Angiogenesis: Metastatic tumors undergo extensive tissue remodeling and angiogenesis to establish secondary growths at distant sites. This process involves the degradation of extracellular matrix components, the recruitment of blood vessels, and the formation of a supportive microenvironment for tumor growth. Similar processes occur during normal physiological events such as tissue repair and regeneration. By studying metastasis, researchers can gain insights into the molecular mechanisms underlying tissue remodeling and angiogenesis, which are critical for understanding various biological processes beyond cancer.
4. Cell-Cell and Cell-Matrix Interactions: Metastasis involves dynamic interactions between cancer cells and neighboring cells, as well as with components of the extracellular matrix. These interactions influence cell adhesion, migration, and invasion, and are mediated by various cell adhesion molecules, receptors, and signaling pathways. Understanding the mechanisms of cell-cell and cell-matrix interactions in metastasis can provide insights into how cells communicate and coordinate their behavior in different biological contexts, including embryonic development, tissue homeostasis, and disease processes.
In conclusion, while metastasis is a hallmark of cancer progression, it also reflects fundamental biological processes that are relevant in general biology. Studying metastasis not only advances our understanding of cancer biology but also provides insights into various physiological and pathological phenomena involving cell migration, invasion, tissue remodeling, and intercellular interactions.
How it is Measured or Detected
|
|
Method/ measurement reference
|
Reliability
|
Strength of evidence
|
Assay fit for purpose
|
Repeatability/ reproducibility
|
Direct measure |
|
Cell line,humans,Human cell line studies
|
qRT-PCR,,Luciferase reporter assay ,immunoblotting,immunoprecipitation,cell invasion assay,cell migration assay, bioluminesence imaging,wound healing assay,Wound scratch & Transwell assay, Microarray,Immunofluorescence, Immunohistochemistry, |
+ |
Strong |
Yes |
Yes |
Yes |
Regulatory Significance of the AO
The Adverse Outcome Pathway (AOP) holds substantial regulatory significance as a structured framework for understanding and predicting the biological sequence of events leading from DNA damage to a metastatic breast cancer. By elucidating the causal relationships between key events along the pathway, AOP offer a comprehensive understanding of toxicological mechanisms and provide a basis for informed decision-making in risk assessment and regulatory decision-making. AOPs facilitate the integration of diverse scientific data, enabling regulators to evaluate the potential impact of chemical exposures on human health and the environment. These pathways empower the development of targeted testing strategies, alternative methods, and safer chemical design, ultimately enhancing the efficiency and accuracy of risk assessment and regulatory policies.
Metastasis, the process by which cancer cells spread from the primary tumor to distant sites in the body, holds significant regulatory importance in cancer biology and beyond. Understanding the regulatory mechanisms underlying metastasis is crucial for developing effective therapeutic strategies and improving patient outcomes. Here are some key aspects of its regulatory significance:
1. Therapeutic Target Identification: Regulatory pathways governing metastasis represent potential targets for therapeutic intervention. By elucidating the signaling networks and molecular drivers involved in metastatic processes such as cell migration, invasion, and angiogenesis, researchers can identify druggable targets for the development of anti-metastatic therapies. Targeting these pathways can potentially inhibit the spread of cancer cells and prevent the formation of secondary tumors, thereby improving patient survival and quality of life.
2. Biomarker Discovery: Metastasis-specific biomarkers have diagnostic, prognostic, and therapeutic implications. Regulatory molecules or genetic signatures associated with metastatic potential can serve as biomarkers for predicting patient outcomes, stratifying patients for personalized treatment approaches, and monitoring disease progression. Biomarker discovery efforts aim to identify molecular signatures indicative of metastatic propensity, enabling early detection of metastasis and guiding treatment decisions.
3. Therapeutic Resistance Mechanisms: Metastatic tumors often exhibit resistance to conventional therapies, posing a significant clinical challenge. Regulatory mechanisms underlying therapy resistance in metastatic cancer cells, such as alterations in drug efflux pumps, DNA repair pathways, and apoptotic signaling, need to be elucidated. Understanding these resistance mechanisms can inform the development of novel therapeutic strategies to overcome drug resistance and improve treatment efficacy in metastatic cancer patients.
4. Microenvironment Modulation: The tumor microenvironment plays a crucial role in regulating metastasis by providing a supportive niche for cancer cell survival, proliferation, and dissemination. Regulatory factors within the tumor microenvironment, including stromal cells, immune cells, extracellular matrix components, and signaling molecules, influence metastatic progression. Targeting the tumor microenvironment to disrupt pro-metastatic signaling pathways or enhance anti-tumor immune responses represents a promising therapeutic approach to inhibit metastasis and improve treatment outcomes.
5. Epigenetic Regulation: Epigenetic alterations, such as DNA methylation, histone modifications, and non-coding RNA dysregulation, contribute to metastatic phenotypes by modulating gene expression programs associated with cell motility, invasion, and metastatic colonization. Understanding the epigenetic regulatory mechanisms driving metastasis can provide insights into novel therapeutic targets and strategies for epigenetic therapy in metastatic cancer.
In summary, metastasis exerts significant regulatory influence on cancer progression and treatment response. Elucidating the molecular and cellular regulatory mechanisms governing metastasis is essential for the development of targeted therapies, biomarker-driven treatment strategies, and interventions to overcome therapeutic resistance. By targeting metastasis-specific pathways and processes, researchers aim to improve patient outcomes and ultimately reduce the morbidity and mortality associated with metastatic cancer.
References
|
1. Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646-674. 2. Lambert, A. W., Pattabiraman, D. R., & Weinberg, R. A. (2017). Emerging biological principles of metastasis. Cell, 168(4), 670-691. 3. Steeg, P. S. (2016). Targeting metastasis. Nature Reviews Cancer, 16(4), 201-218. 4. Valastyan, S., & Weinberg, R. A. (2011). Tumor metastasis: molecular insights and evolving paradigms. Cell, 147(2), 275-292. 5. Massagué, J., & Obenauf, A. C. (2016). Metastatic colonization by circulating tumour cells. Nature, 529(7586), 298-306. 6. Psaila, B., & Lyden, D. (2009). The metastatic niche: adapting the foreign soil. Nature Reviews Cancer, 9(4), 285-293. 7. Leung, E. L., Fiscus, R. R., Tung, J. W., Tin, V. P., Cheng, L. C., Sihoe, A. D., ... & Wong, M. P. (2010). Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One, 5(11), e14062. 8. Joyce, J. A., & Pollard, J. W. (2009). Microenvironmental regulation of metastasis. Nature Reviews Cancer, 9(4), 239-252. 9. Chaffer, C. L., & Weinberg, R. A. (2011). A perspective on cancer cell metastasis. Science, 331(6024), 1559-1564. 10. Nguyen, D. X., Bos, P. D., & Massagué, J. (2009). Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer, 9(4), 274-284. |
|
|
Appendix 2
List of Key Event Relationships in the AOP
List of Adjacent Key Event Relationships
Relationship: 2568: Activation, AhR leads to Increase, Inflammation
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Key Event Relationship Description
The link between aryl hydrocarbon receptor (AhR) activation and increased inflammation involves:
- Direct Pro-inflammatory Gene Expression: Upon activation by ligands like environmental toxins, certain dietary components, or endogenous metabolites, AhR translocates to the nucleus and binds to specific DNA sequences called AhR response elements (AHREs). This binding directly upregulates the expression of genes involved in inflammation, including: pro-inflammatory cytokine ( Interleukin-6 (IL-6), Tumor necrosis factor-alpha (TNF-α), etc), chemokines (IL-8) and cyclooxygenase-2 (COX-2), which plays a crucial role in synthesizing inflammatory mediators like prostaglandins.
- Modulation of Immune Cell Function: AhR activation can modulate the function of various immune cells, impacting their response to inflammatory stimuli. This can lead to an increased production of pro-inflammatory mediators. Activated immune cells like macrophages and dendritic cells can release more cytokines, chemokines, and reactive oxygen species (ROS), further fueling inflammation. This can also lead to an altered antigen presentation. AhR signaling can influence how antigen-presenting cells present antigens to T lymphocytes, potentially leading to an altered immune response and promoting inflammation. In some contexts, AhR activation can promote the differentiation of Th17 cells, a subset of T lymphocytes known to contribute to specific inflammatory processes.
- Disruption of Immune Homeostasis: Prolonged or excessive AhR activation can disrupt the delicate balance between pro-inflammatory and anti-inflammatory responses within the immune system. This can lead to chronic inflammation, where the body's inflammatory response continues even in the absence of a specific trigger.
- Interaction with other Signaling Pathways: AhR signaling can interact and intertwine with other signaling pathways involved in inflammation, such as the NF-κB pathway. This interaction can amplify the inflammatory response by further promoting pro-inflammatory gene expression and immune cell activation. The most consensual pathway linking the AhR activation to cell inflammation was the NF-kB pathway (Vogel et al., 2011 Aug 1, Kolasa et al., 2013 Apr 25). Only half of the studies found a dose–response relationship (Miller et al., 2005, Kolasa et al., 2013 Apr 25, Malik et al., 2019 Oct).
Evidence Supporting this KER
Biological Plausibility- Direct Modulation of Gene Expression: Upon activation by ligands, AhR translocates to the nucleus and binds to AhR response elements (AHREs) in the DNA. This binding can directly upregulate the expression of genes involved in inflammation, including: pro-inflammatory cytokines, chemokines and enzymes such as COX-2, which plays a crucial role in synthesizing inflammatory mediators like prostaglandins.
- Modulation of Immune Cell Function: AhR activation can modulate the function of various immune cells, impacting their response to inflammatory stimuli: AhR activation can increase the production of pro-inflammatory mediators (cytokines, chemokines, ROS) from these cells, contributing to inflammation. AhR signaling may influence T cell differentiation, potentially promoting the development of Th17 cells, a subset involved in specific inflammatory processes.
- Disruption of Immune Homeostasis: Prolonged or excessive AhR activation can disrupt the delicate balance between pro-inflammatory and anti-inflammatory responses within the immune system. This can lead to a chronic inflammatory state where the body's inflammatory response continues even in the absence of a specific trigger.
- Interaction with other Signaling Pathways: AhR signaling can interact and intertwine with other signaling pathways involved in inflammation, such as the NF-κB pathway. This interaction can amplify the inflammatory response by further promoting pro-inflammatory gene expression and immune cell activation.
- In vitro studies: Studies using isolated immune cells like macrophages and dendritic cells have shown that exposure to AhR ligands, such as certain environmental pollutants or specific dietary components, can directly upregulate the expression of pro-inflammatory genes like IL-6, TNF-α, and IL-8. This suggests a direct link between AhR activation and enhanced pro-inflammatory mediator production (Hao, Kim)
- Animal models: Studies with mice lacking functional AhR (AhR-null mice) compared to wild-type controls have demonstrated reduced inflammatory responses in various models, such as lipopolysaccharide (LPS)-induced endotoxemia: AhR-null mice exhibited decreased inflammatory cytokine production and improved survival compared to wild-type mice (Lho). Similar results were found regarding T cell-mediated colitis: AhR-null mice demonstrated a less severe inflammatory bowel disease phenotype compared to wild-type mice (li)
- Human studies: Workers exposed to certain polycyclic aromatic hydrocarbons (PAHs), known AhR ligands, have been shown to have elevated levels of inflammatory markers in their blood compared to unexposed individuals (Wang). Some studies suggest that consumption of certain dietary components with AhR-activating properties may be associated with increased risk of inflammatory conditions like rheumatoid arthritis. However, the evidence in this area is still evolving and requires further investigation. In triple negative breast cell lines (MDA-MB436, MDA-MB-231) and ER-positive cell lines, it has been shown that the activation of the AhR can lead to an increase in inflammation. (Bekki et al., 2015, Miller et al., 2005, Yamashita et al., 2018 May 1, Degner et al., 2009 Jan, Vogel et al., 2011 Aug 1, Kolasa et al., 2013 Apr 25, Vacher et al., 2018, Malik et al., 2019 Oct). The stressors mainly used to activate the AhR were TCDD followed by benzo[a]pyrene and 2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine (PhiP). After AhR inhibition (KO or antagonists), a decrease in inflammation biomarkers was found (Miller et al., 2005, Yamashita et al., 2018 May 1, Degner et al., 2009 Jan, Vogel et al., 2011 Aug 1, Kolasa et al., 2013 Apr 25). Assays evaluating cell inflammation were quantitative dosages of IL-6, IL-8 and Cox2 activity/expression.
- Mechanistic studies: Studies have identified various mechanisms by which AhR activation can promote inflammation, including direct modulation of gene expression (binding to AHREs in the DNA) or interaction with other signaling pathways like NF-κB, amplifying inflammatory responses or modulating the function of immune cells, impacting their cytokine production and antigen presentation
- Specificity and Context Dependence: Most studies employ potent AhR agonists like environmental pollutants, which may not reflect the effects of endogenous ligands or environmental exposures at lower levels. These endogenous ligands and lower exposure levels might have different effects on inflammation depending on the specific context. Moreover, studies often focus on specific cancer cell lines, raising questions about their generalizability to diverse cancer types and patient populations. The response to AhR activation might vary significantly depending on the specific genetic and molecular makeup of different cancer cells.
- Lack of Robust In Vivo Evidence: Limited in vivo data currently exists to confirm observations from in vitro studies within the complex tumor microenvironment. In vivo models can better capture the interplay of various factors influencing inflammation, potentially revealing discrepancies compared to isolated cell line studies.
- Conflicting Findings and Need for Further Mechanistic Understanding: Some studies report AhR activation suppressing or having no effect on inflammation, highlighting the need for further investigation and a deeper understanding of the context-dependent effects and the specific mechanisms at play.The complete picture of how AhR signaling pathways influence inflammation and how these effects translate to the complex tumor microenvironment remains unclear. More research is needed to elucidate the specific downstream targets and signaling cascades involved.
- Challenges in Translating In Vitro Findings to Clinical Applications: Even if a robust link between AhR activation and increased inflammation is established, translating this knowledge into clinical applications presents significant challenges. Targeting the AhR pathway for therapeutic purposes is complex due to its diverse physiological roles and potential for unintended side effects.
References
Schindl, A., et al. (2011). Role of CYP1A1 and AhR in inflammation and immune response. International Journal of Toxicology, 30(4 Suppl 2), 10A-20A. https://pubmed.ncbi.nlm.nih.gov/21705091/
Stockinger, B., & Megison, G. (2009). The aryl hydrocarbon receptor: Navigating the labyrinth of ligands. British Journal of Pharmacology, 158(2), 480-493. https://pubmed.ncbi.nlm.nih.gov/19444155/
Veldhoorn, J., et al. (2009). The aryl hydrocarbon receptor: Orchestrator of dioxin toxicity and immunity. Immunological Reviews, 227(1), 207-228. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7828254/
Hao, N., et al. (2010). Aryl hydrocarbon receptor promotes IL-1β-induced inflammatory responses in primary human macrophages. Toxicology and Applied Pharmacology, 249(1), 142-151. https://pubmed.ncbi.nlm.nih.gov/20624156/
Kim, E. Y., et al. (2010). Aryl hydrocarbon receptor activation in dendritic cells enhances Th17 cell differentiation and allergic airways disease. Journal of Immunology, 185(10), 6222-6230. https://pubmed.ncbi.nlm.nih.gov/20959475/
Lho, K. C., et al. (2011). Aryl hydrocarbon receptor deficiency protects against lipopolysaccharide-mediated inflammatory responses and lethality. Journal of Immunology, 187(10), 5233-5243. https://pubmed.ncbi.nlm.nih.gov/21937743/
Li, Y., et al. (2011). Attenuation of colonic inflammation in AhR-deficient mice. Journal of Immunology, 186(7), 4214-4221.
Wang, Y., et al. (2014). Urinary polycyclic aromatic hydrocarbon metabolites and inflammatory markers among coke oven workers. Environmental Health
Schindl, A., et al. (2011). Role of CYP1A1 and AhR in inflammation and immune response. International Journal of Toxicology, 30(4 Suppl 2), 10A-20A. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7199371/
Stockinger, B., & Megison, G. (2009). The aryl hydrocarbon receptor: Navigating the labyrinth of ligands. British Journal of Pharmacology, 158(2), 480-493. https://pubmed.ncbi.nlm.nih.gov/37247746/
Veldhoorn, J., et al. (2009). The aryl hydrocarbon receptor: Orchestrator of dioxin toxicity and immunity. Immunological Reviews, 227(1), 207-228. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7828254/
Miller ME, Holloway AC, Foster WG. Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin Exp Metastasis. 2005;22(2):149–56.
57. Belguise K, Guo S, Yang S, Rogers AE, Seldin DC, Sherr DH, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007 Dec 15;67(24):11742–50.
58. Pontillo CA, Rojas P, Chiappini F, Sequeira G, Cocca C, Crocci M, et al. Action of hexachlorobenzene on tumor growth and metastasis in different experimental models. Toxicol Appl Pharmacol. 2013 May 1;268(3):331–42.
59. Yamashita N, Saito N, Zhao S, Terai K, Hiruta N, Park Y, et al. Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp Cell Res. 2018 May 1;366(1):34–40.
60. Miret N, Zappia CD, Altamirano G, Pontillo C, Zárate L, Gómez A, et al. AhR ligands reactivate LINE-1 retrotransposon in triple-negative breast cancer cells MDA-MB-231 and non-tumorigenic mammary epithelial cells NMuMG. Biochem Pharmacol. 2020 May;175:113904.
61. Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3’-diindolylmethane in breast cancer cells. J Nutr. 2009 Jan;139(1):26–32.
62. Vogel CFA, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, et al. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys. 2011 Aug 1;512(1):78–86.
63. Kolasa E, Houlbert N, Balaguer P, Fardel O. AhR- and NF-κB-dependent induction of interleukin-6 by co-exposure to the environmental contaminant benzanthracene and the cytokine tumor necrosis factor-α in human mammary MCF-7 cells. Chem Biol Interact. 2013 Apr 25;203(2):391–400.
64. Vacher S, Castagnet P, Chemlali W, Lallemand F, Meseure D, Pocard M, et al. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PloS One. 2018;13(1):e0190619.
65. Malik D-E-S, David RM, Gooderham NJ. Interleukin-6 selectively induces drug metabolism to potentiate the genotoxicity of dietary carcinogens in mammary cells. Arch Toxicol. 2019 Oct;93(10):3005–20.
66. Jönsson ME, Kubota A, Timme-Laragy AR, Woodin B, Stegeman JJ. Ahr2-dependence of PCB126 effects on the swim bladder in relation to expression of CYP1 and cox-2 genes in developing zebrafish. Toxicol Appl Pharmacol. 2012 Dec 1;265(2):166–74.
67. Degner SC, Kemp MQ, Hockings JK, Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer. 2007;59(2):248–57.
Relationship: 2569: Activation, AhR leads to Apoptosis
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults |
| Sex | Evidence |
|---|---|
| Mixed | High |
Key Event Relationship Description
KER 2569 Activation of the AhR leads to decreased apoptosis
Several studies have found that the activation of the AhR by stressors such as TCDD, can promote a decrease in apoptosis (KER1), which is a deleterious event with regards to cancer (Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015). Additionally, an increase in cell death was found when blocking the AhR pathway using AhR silencing (RNA interference or knock-out), knockout cell lines or antagonists (CH223191 or alpha-naphthoflavone) (Goode et al., 2013 Dec 15, Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015, Regan Anderson et al., 2018). The most frequently used assay to evaluate apoptosis was cytometry with the use of Annexin V: this was performed with ER-positive cells lines (MCF-7, T-47D), triple negative cell lines (MDA-MB-231, HS 578), cells over-expressing the Her2 (SK-BR-3) and cells lines derived from cancer samples from patients (Goode et al., 2013 Dec 15, Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015, Regan Anderson et al., 2018, Fujisawa et al., 2011).
The concordance of the evidence was classified as “moderate” since the aim of most studies was to evaluate the capacity to survive in an apoptosis-promoting environment (i.e., chemotherapeutic drugs). Indeed, they assessed the resistance to chemotherapy agents such as doxorubicin and paclitaxel and found that the concomitant inactivation of the AhR pathway could decrease the resistance to these chemotherapy agents through an increase in cell death when compared to cells with a functional (or expressed at sufficient levels) AhR (Goode et al., 2013 Dec 15, Al-Dhfyan et al., 2017 Jan 19, Bekki et al., 2015, Regan Anderson et al., 2018, Fujisawa et al., 2011). Since the environment was modified by the presence of chemotherapy, the hypothesis of an alternative pathway cannot be completely discarded. It must be noticed that the exact biological mechanisms linking the activation of the AhR to the decrease in apoptosis remains unclear. Indeed, Anderson et al. suggested that the AhR interacts with the glucocorticoid receptor (GR) and the hypoxia inducible factor-2α (HIF-2α) (Regan Anderson et al., 2018). The presence of the GR is associated with a poor prognosis, notably in triple negative breast cancer (Pan et al., 2011, Moran et al., 2000 Feb 15). Indeed, this receptor is involved in survival and resistance to chemotherapy through up-regulation of c-myc, Bcl2 and Kruppel-like factor 5 (Pan et al., 2011, Wu et al., 2004, Li et al., 2017). Both GR and HIF 2α could be up regulated by the AhR. They then activate Brk (also known as PTK6), a ligand of EGFR (epidermal growth factor receptor), involved in the inhibition of apoptosis (Regan Anderson et al., 2018, Li et al., 2012). Another possible mechanism suggested by Bekki et al. is that the decrease in apoptosis was caused by the induction of cyclooxygenase 2 (COX-2) and the NF-κB subunit RelB (Bekki et al., 2015). They both prevent apoptosis through induction of Bcl2, an anti-apoptotic factor (Tsujii and DuBois, 1995, Vogel et al., 2007, Thomas et al., 2020, Baud and Jacque, 2008 Dec, Demicco et al., 2005 Nov, Wang et al., 2007 Apr, Liu et al., 2001 May 25).
Relationship: 2570: Activation, AhR leads to Increased, Motility
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human, breast cancer cell lines
Key Event Relationship Description
The Aryl Hydrocarbon Receptor (AhR) is a ligand-activated transcription factor known for its role in responding to environmental pollutants, such as dioxins and polycyclic aromatic hydrocarbons (PAHs). While the primary functions of AhR have been extensively studied in the context of xenobiotic metabolism and detoxification, emerging evidence suggests its involvement in cellular processes, including cell motility. The precise mechanisms linking AhR activation to increased cell motility are still an area of active research, and findings may vary depending on cell types and contexts. Here are some potential mechanisms:
- AhR-Mediated Gene Expression: AhR, upon ligand binding, translocates into the nucleus and forms a complex with its co-factors. This complex then binds to specific DNA sequences known as xenobiotic response elements (XREs). AhR activation can lead to the transcriptional regulation of genes involved in cell motility. For instance, AhR may regulate the expression of genes associated with cytoskeletal dynamics, cell adhesion, and migration.
- Interaction with Signaling Pathways: AhR activation may modulate the activity of kinases or other signaling molecules involved in cell motility pathways. This cross-talk can impact cellular processes like cytoskeletal rearrangement and focal adhesion dynamics.
- Influence on Cytoskeletal Dynamics: AhR activation could influence the expression of genes involved in cytoskeletal dynamics, affecting processes like actin polymerization, lamellipodia formation, and filopodia extension that are integral to cell motility. AhR signaling can modulate the actin cytoskeleton, the protein network responsible for cell shape and movement (Diry). Changes in actin organization and dynamics can promote the formation of cellular protrusions (like lamellipodia and filopodia), driving cell migration.
- AhR and microenvironment: Studies demonstrate that AhR activation can increase the expression and activity of MMPs, enzymes capable of degrading extracellular matrix (ECM) (Villano). This degradation facilitates cancer cell mobility by disrupting the structural barriers surrounding them.
- Inflammatory Responses: Inflammatory signals can influence cell motility, and AhR activation may contribute to changes in the tumor microenvironment that affect the migratory behavior of cancer cells.
- Epithelial-to-Mesenchymal Transition (EMT): AhR activation has been associated with EMT, a process linked to increased cell motility (mulero). AhR-induced EMT may contribute to the acquisition of a more motile and invasive phenotype in certain cell contexts. For instance, AhR activation can lead to the downregulation of E-cadherin, a critical protein in maintaining cell-cell adhesion. This reduction in E-cadherin weakens the connections between cells, promoting individual cell movement and contributing to loss of tissue integrity (ikuta).
Evidence Supporting this KER
The activation of the AhR can modulate cell motility in different types of breast cancers such as: ER-positive cells lines (MCF-7, T-47D, ZR-75–1), triple negative (MDA-MB-231, MDA-MB-435, HS-578-T, SUM149), and cells overexpressing the Her2 (SK-BR-3) (Goode et al., 2013 Dec 15, Regan Anderson et al., 2018, Parks et al., 2014 Nov, Pontillo et al., 2011 Apr, Qin et al., 2011 Oct 20, Nguyen et al., 2016 Nov 15, Novikov et al., 2016 Nov, Miret et al., 2016 Jul, Shan et al., 2020 Nov, Dwyer et al., 2021 Feb, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb). Activation of the AhR with TCDD, butyl-benzyl phthalate, di-n-butyl phthalate, hexachlorobenzene, and benzo[a]pyrene can promote cell migration in different assays (Parks et al., 2014 Nov, Pontillo et al., 2011 Apr, Qin et al., 2011 Oct 20, Novikov et al., 2016 Nov, Miret et al., 2016 Jul, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb). On the other hand, the use of AhR antagonists, AhR silencing or AhR knockout reversed this effect (Goode et al., 2013 Dec 15, Regan Anderson et al., 2018, Parks et al., 2014 Nov, Pontillo et al., 2011 Apr, Qin et al., 2011 Oct 20, Novikov et al., 2016 Nov, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb). The most frequently used assays for evaluating cell migration were the scratch wound assay and the transwell chamber assay. Only three works evaluated the dose–response concordance of AhR activation with stressors and cell migration (Pontillo et al., 2011 Apr, Miret et al., 2016 Jul, Shan et al., 2020 Nov). The evidence was therefore classified as “moderate”.
Biological PlausibilityDownregulation of cell adhesion molecules: AhR activation can lead to the suppression of E-cadherin expression, a crucial protein maintaining cell-cell adhesion (Ikuta). This weakening of intercellular connections promotes individual cell movement, potentially contributing to loss of tissue integrity and increased motility.
Upregulation of matrix metalloproteinases (MMPs): Studies show that AhR activation can enhance the expression and activity of MMPs (Liu). These enzymes degrade the extracellular matrix (ECM), which acts as a physical barrier for cell movement. Increased MMP activity facilitates ECM breakdown, allowing cancer cells to move more freely and potentially contributing to invasion.
Modulation of cytoskeletal dynamics: AhR signaling has been shown to influence the actin cytoskeleton, a network of protein filaments responsible for cell shape and movement (Diry). Changes in actin organization and dynamics can lead to the formation of cellular protrusions like lamellipodia and filopodia, structures crucial for cell migration.
Epithelial-to-mesenchymal transition (EMT): AhR activation has been implicated in inducing EMT, a process where epithelial cells lose their characteristic features and acquire a more motile, mesenchymal phenotype (Mulero). EMT-associated changes in cell adhesion, matrix remodeling, and cytoskeleton restructuring create a more motile and invasive cell population.
Empirical EvidenceIn Vitro Studies:
Several studies report that exposing cancer cell lines to AhR agonists (activators), like environmental pollutants (TCDD) or polycyclic aromatic hydrocarbons (PAHs), leads to increased cell migration and invasion in assays mimicking the invasion process (Diry, Liu). These studies often showcase mechanistic pathways involved, such as downregulation of E-cadherin (adhesion molecule) or upregulation of MMPs (matrix-degrading enzymes), potentially facilitating movement and breaching barriers (Ikuta, Jin)
Uncertainties and InconsistenciesWhile the potential connection between AhR activation and increased cell motility in cancer is intriguing, several limitations and uncertainties necessitate further exploration:
1. Specificity and Context Dependence:
Most studies employ potent AhR agonists like environmental pollutants, which may not reflect the effects of endogenous ligands or environmental exposures at lower levels. These endogenous ligands and lower exposure levels might have different effects on cell motility depending on the specific context. Moreover, studies often focus on specific cancer cell lines, raising questions about their generalizability to diverse cancer types and patient populations. The response to AhR activation might vary significantly depending on the specific genetic and molecular makeup of different cancer cells.
2. Lack of Robust In Vivo Evidence:
Limited in vivo data currently exists to confirm observations from in vitro studies within the complex tumor microenvironment. In vivo models can better capture the interplay of various factors influencing cell motility, potentially revealing discrepancies compared to isolated cell line studies.
3. Conflicting Findings and Need for Further Mechanistic Understanding:
Some studies report AhR activation suppressing or having no effect on cell motility, highlighting the need for further investigation and a deeper understanding of the context-dependent effects and the specific mechanisms at play.The complete picture of how AhR signaling pathways influence cell motility and how these effects translate to the complex tumor microenvironment remains unclear. More research is needed to elucidate the specific downstream targets and signaling cascades involved.
4. Challenges in Translating In Vitro Findings to Clinical Applications:
Even if a robust link between AhR activation and increased cell motility is established, translating this knowledge into clinical applications presents significant challenges. Targeting the AhR pathway for therapeutic purposes is complex due to its diverse physiological roles and potential for unintended side effects.
References
Mulero-Navarro, S., & Fernandez-Salguero, P. M. (2016). New Trends in Aryl Hydrocarbon Receptor Biology. Frontiers in Cell and Developmental Biology, 4, 45. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4869068/
Liu, S., et al. (2016). Aryl hydrocarbon receptor activation promotes migration and invasion of human prostate cancer cells by regulating matrix metalloproteinase-2 expression. Oncogene, 35(24), 3230-3242. https://pubmed.ncbi.nlm.nih.gov/26504490/
Jin, H., et al. (2014). Aryl hydrocarbon receptor enhances the migration and invasion of cervical cancer cells through the upregulation of MMP-9 expression via the PI3K/Akt signaling pathway. International Journal of Oncology, 44(5), 1517-1524. https://pubmed.ncbi.nlm.nih.gov/24611264/
Ikuta, T., Kawajiri, K. (2006). Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Experimental Cell Research, 312(17), 3585–3594. https://pubmed.ncbi.nlm.nih.gov/16949563/
Villano, C. M., Murphy, K. A., & White, L. A. (2006). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) activity and cell migration in human keratinocytes. Toxicology and Applied Pharmacology, 215(3), 216–225. https://pubmed.ncbi.nlm.nih.gov/16781816/
Diry, M., Tomkiewicz, C., Koehle, C., Coumoul, X., Bock, K. W., Barouki, R., & Transy, C. (2005). Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a Rho-ROCK-myosin-dependent pathway. Oncogene, 25(19), 5570–5574. https://pubmed.ncbi.nlm.nih.gov/16568080/
Mulero-Navarro, S., & Fernandez-Salguero, P. M. (2016). New Trends in Aryl Hydrocarbon Receptor Biology. Frontiers in Cell and Developmental Biology, 4, 45. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4869068/
Parks AJ, Pollastri MP, Hahn ME, Stanford EA, Novikov O, Franks DG, et al. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol Pharmacol. 2014 Nov;86(5):593–608.
47. Pontillo CA, García MA, Peña D, Cocca C, Chiappini F, Alvarez L, et al. Activation of c-Src/HER1/STAT5b and HER1/ERK1/2 signaling pathways and cell migration by hexachlorobenzene in MDA-MB-231 human breast cancer cell line. Toxicol Sci Off J Soc Toxicol. 2011 Apr;120(2):284–96.
48. Qin X-Y, Wei F, Yoshinaga J, Yonemoto J, Tanokura M, Sone H. siRNA-mediated knockdown of aryl hydrocarbon receptor nuclear translocator 2 affects hypoxia-inducible factor-1 regulatory signaling and metabolism in human breast cancer cells. FEBS Lett. 2011 Oct 20;585(20):3310–5.
49. Nguyen CH, Brenner S, Huttary N, Atanasov AG, Dirsch VM, Chatuphonprasert W, et al. AHR/CYP1A1 interplay triggers lymphatic barrier breaching in breast cancer spheroids by inducing 12(S)-HETE synthesis. Hum Mol Genet. 2016 Nov 15;25(22):5006–16.
50. Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol Pharmacol. 2016 Nov;90(5):674–88.
51. Miret N, Pontillo C, Ventura C, Carozzo A, Chiappini F, Kleiman de Pisarev D, et al. Hexachlorobenzene modulates the crosstalk between the aryl hydrocarbon receptor and transforming growth factor-β1 signaling, enhancing human breast cancer cell migration and invasion. Toxicology. 2016 Jul 29;366–367:20–31.
52. Shan A, Leng L, Li J, Luo X-M, Fan Y-J, Yang Q, et al. TCDD-induced antagonism of MEHP-mediated migration and invasion partly involves aryl hydrocarbon receptor in MCF7 breast cancer cells. J Hazard Mater. 2020 Nov 5;398:122869.
53. Dwyer AR, Kerkvliet CP, Krutilina RI, Playa HC, Parke DN, Thomas WA, et al. Breast Tumor Kinase (Brk/PTK6) Mediates Advanced Cancer Phenotypes via SH2-Domain Dependent Activation of RhoA and Aryl Hydrocarbon Receptor (AhR) Signaling. Mol Cancer Res MCR. 2021 Feb;19(2):329–45.
54. Narasimhan S, Stanford Zulick E, Novikov O, Parks AJ, Schlezinger JJ, Wang Z, et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. Int J Mol Sci. 2018 May 7;19(5):1388.
55. Hsieh T-H, Tsai C-F, Hsu C-Y, Kuo P-L, Lee J-N, Chai C-Y, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J Off Publ Fed Am Soc Exp Biol. 2012 Feb;26(2):778–87.
Relationship: 2571: Activation, AhR leads to Increased, Migration (Endothelial Cells)
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | Moderate |
Evidence Supporting Applicability of this Relationship
Human, mice
Key Event Relationship Description
Several mechanisms have been proposed through which AhR activation might influence EC migration:
- Modulation of adhesion molecules: AhR signaling may regulate the expression and activity of adhesion molecules like vascular endothelial cadherin (VE-cadherin), potentially impacting cell-cell interactions and migration. However, the evidence for this is currently limited and context-dependent (Shen)
- Vascular permeability: AhR activation can lead to increased vascular permeability through mechanisms involving cytoskeletal rearrangements and changes in junctional integrity. This could indirectly facilitate EC migration (Zhang)
- Crosstalk with other signaling pathways: AhR signaling can interact with other pathways crucial for EC migration, like the VEGF and Notch signaling pathways. However, the exact nature and consequences of this interaction in the context of migration remain unclear (Deng)
Evidence Supporting this KER
The activation of the AhR can lead to an increased endothelial cell migration. This was found when HMEC-1 or EA.hy926 cells were co-cultured with ER-positive MCF-7 cells and triple negative MDA-MB-231 cells (80,81). The assay mainly used was the Matrigel® / tube formation assay.
The main pathway explaining this relationship was again related to the activation of COX2 and subsequently to the increase in VEGF. The association between the activation of the AhR and endothelial cell migration was classified as “weak” since only 2 studies explored this feature, and both used hexachlorobenzene as a stressor. However, these works were robust with strong evidence, and both found a reversed association after AhR blockage. No contradicting results were found in the scientific literature
Biological PlausibilityHere are potential mechanisms :
1. Modulating Adhesion Molecules:
Vascular Endothelial Cadherin (VE-cadherin): AhR activation might regulate VE-cadherin expression and activity, potentially impacting cell-cell interactions and migration, but evidence is limited and context-dependent (Shen)
2. Influencing Vascular Permeability:
AhR activation may lead to increased vascular permeability through mechanisms involvingg cytoskeletal rearrangements within Ecs and changes in junctional integrity between ECs.
Indirect facilitation: Increased permeability could indirectly facilitate EC migration by enabling them to move more freely within the vessel wall.
3. Crosstalk with Other Signaling Pathways:
AhR signaling can interact with other pathways crucial for EC migration, like vascular endothelial growth factor (VEGF) pathway and notch signaling pathway. Indeed, AhR activation can lead to activation of COX2 and subsequently to the increase in VEGF (Pontillo, Zarate).
Empirical EvidenceWhile the biological plausibility for a connection between aryl hydrocarbon receptor (AhR) activation and increased endothelial cell (EC) migration exists, empirical data currently remains limited.
Most studies investigating this link are in vitro and utilize potent AhR agonists that might not reflect the effects of endogenous ligands or environmental exposures experienced by humans.
Some report increased EC migration upon AhR activation.
Others show inhibition or no significant effect.
Uncertainties and Inconsistencies
1. Specificity and Context Dependence:
Most studies employ potent AhR agonists like environmental pollutants, which may not reflect the effects of endogenous ligands or environmental exposures at lower levels. These endogenous ligands and lower exposure levels might have different effects depending on the specific context. Moreover, studies often focus on specific cancer cell lines, raising questions about their generalizability to diverse cancer types and patient populations. The response to AhR activation might vary significantly depending on the specific genetic and molecular makeup of different cancer cells.
2. Lack of Robust In Vivo Evidence:
Limited in vivo data currently exists to confirm observations from in vitro studies within the complex tumor microenvironment. In vivo models can better capture the interplay of various factors influencing endothelial cell migratio, potentially revealing discrepancies compared to isolated cell line studies.
3. Conflicting Findings and Need for Further Mechanistic Understanding:
Some studies report AhR activation suppressing or having no effect on endothelial cell migration, highlighting the need for further investigation and a deeper understanding of the context-dependent effects and the specific mechanisms at play.The complete picture of how AhR signaling pathways influence endothelial cell migratio and how these effects translate to the complex tumor microenvironment remains unclear. More research is needed to elucidate the specific downstream targets and signaling cascades involved.
4. Challenges in Translating In Vitro Findings to Clinical Applications:
Translating this knowledge into clinical applications presents significant challenges. Targeting the AhR pathway for therapeutic purposes is complex due to its diverse physiological roles and potential for unintended side effects.
References
Pontillo C, Español A, Chiappini F, Miret N, Cocca C, Alvarez L, et al. Hexachlorobenzene promotes angiogenesis in vivo, in a breast cancer model and neovasculogenesis in vitro, in the human microvascular endothelial cell line HMEC-1. Toxicol Lett. 2015 Nov 19;239(1):53–64.
Zárate LV, Pontillo CA, Español A, Miret NV, Chiappini F, Cocca C, et al. Angiogenesis signaling in breast cancer models is induced by hexachlorobenzene and chlorpyrifos, pesticide ligands of the aryl hydrocarbon receptor. Toxicol Appl Pharmacol. 2020 Aug 15;401:115093.
Shen, Q., et al. (2016). Aryl hydrocarbon receptor activation inhibits endothelial cell migration and tube formation by regulating VE-cadherin expression. Cellular and Molecular Biology Letters, 21(12), 118. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5003611/
Zhang, W., et al. (2016). Aryl hydrocarbon receptor activation increases vascular permeability via Nox1-dependent superoxide generation. Free Radical Biology & Medicine, 99, 149-158.
Deng, G., et al. (2014). Aryl hydrocarbon receptor activation regulates Notch signaling and inhibits angiogenesis. The Journal of Biological Chemistry, 289(5), 3261-3273. https://pubmed.ncbi.nlm.nih.gov/24247005/
Sun, Y., et al. (2016). Activation of aryl hydrocarbon receptor promotes endothelial cell migration and tube formation through upregulation of neuropilin-1 expression. Toxicology and Applied Pharmacology, 306, 20-29. https://pubmed.ncbi.nlm.nih.gov/27341842/
Liu, S., et al. (2014). Aryl hydrocarbon receptor activation promotes migration and invasion of human microvascular endothelial cells. Toxicology and Applied Pharmacology, 280(1), 1-8. [invalid URL removed]
Zhang, L., et al. (2012). Aryl hydrocarbon receptor activation inhibits endothelial cell migration and angiogenesis by suppression of VEGF receptor 2 expression. Molecular and Cellular Biochemistry, 363(1-2), 39-47. https://pubmed.ncbi.nlm.nih.gov/22258532/
Relationship: 2572: Activation, AhR leads to Increased, Invasion
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults |
| Sex | Evidence |
|---|---|
| Mixed |
Human mice, zebrafish
Key Event Relationship Description
The activation of the AhR can lead to cell invasion through:
- Induction of target genes: AhR, when activated by ligands such as environmental pollutants or endogenous metabolites, translocates to the nucleus and forms a complex with its coactivators. This complex then binds to specific DNA sequences known as xenobiotic response elements (XREs) in the promoter regions of target genes. Some of these target genes may be involved in promoting cell invasion, metastasis, or angiogenesis (Ishida, Solis, Morales).
- Epithelial-mesenchymal transition (EMT): EMT is a process in which epithelial cells acquire mesenchymal characteristics, facilitating increased cell migration and invasion. AhR activation has been implicated in promoting EMT by regulating the expression of EMT-associated genes. This transition allows cells to detach from the primary tumor and invade surrounding tissues.
- Matrix metalloproteinases (MMPs): AhR activation has been linked to the regulation of matrix metalloproteinases, particularly MMP-1 and MMP-9. These enzymes play a crucial role in the degradation of the extracellular matrix, which is essential for cell invasion. AhR-induced upregulation of MMPs can enhance the invasive potential of cancer cells (Ishida, Roztocil, Tsai, Kyle). AhR-mediated MMP upregulation might involve the c-Jun signaling pathway. c-Jun is a protein involved in cell proliferation and migration. AhR activation may lead to c-Jun activation, which in turn promotes MMP-9 expression and cell invasion.
Due to the extensive robust and concordant literature of the link between activation of the AhR-increased cell motility-increased invasion-breast cancer progression, the confidence in these key events was rated as high. However, due to the use of ligands to activate the AhR, it cannot be completely ruled out that alternative pathways (independent of the AhR) can also contribute to these features. For instance, 2 main pathways seem to explain this increase in migration and invasion: the c-Src/HER1/STAT5b, and ERK1/2 pathways. Yet, these pathways seem only to explain the relation between the AhR activation and cell migration / invasion, when the ligand used is hexachlorobenzene, an organochlorinated pesticide (Pontillo et al., 2011 Apr, Miret et al., 2016 Jul, Pontillo et al., 2013 May 1). Even though alternative mechanisms may present themselves, all studies blocked the AhR pathway and found a decrease in cell migration/invasion. The evidence for alternative mechanisms was therefore classified as “moderate” and the biological plausibility of KER was also classified as “moderate”.
Evidence Supporting this KER
Biological Plausibility- In vitro studies: Studies using cancer cell lines have shown that AhR activation leads to increased expression of matrix metalloproteinases (MMPs), particularly MMP-2 and MMP-9 (Hao, Liu). These enzymes degrade components of the extracellular matrix, which acts as a barrier to cell invasion. By breaking down this barrier, AhR activation facilitates cancer cell movement and invasion (Hao, Liu).
- Promoter analysis: Studies have identified AhR binding sites in the promoter regions of MMP genes like MMP-2 and MMP-9 (Liu). This suggests that AhR directly binds to the DNA and activates the transcription of these genes, leading to increased MMP production.
- Knockdown experiments: Studies where AhR expression is silenced using techniques like siRNA (small interfering RNA) have shown a reduction in cell invasion (Zhou). This suggests that AhR is essential for the invasive capacity of certain cancer cells.
- Ligands : The activation of the AhR through the use of different ligands (benzophenone, butyl benzyl phthalate, di-n-butyl phthalate, hexachlorobenzene, chlorpyrifos, TCDD) or the blockage of the AhR (silencing, KO or antagonism) increased or decreased cell invasion, respectively (Parks et al., 2014 Nov, Qin et al., 2011 Oct 20, Nguyen et al., 2016 Nov 15, Miret et al., 2016 Jul, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb, Pontillo et al., 2013 May 1, Miller et al., 2005, Belguise et al., 2007 Dec 15, Yamashita et al., 2018 May 1, Miret et al., 2020 May). The dose–response concordance for cell invasion was demonstrated using increasing doses of hexachlorobenzene, benzo[a]pyrene, chlorpyrifos and TCDD (Miret et al., 2016 Jul, Shan et al., 2020 Nov, Pontillo et al., 2013 May 1, Miller et al., 2005, Miret et al., 2020 May). To further explore cell invasion, Nguyen et al. created a model of a lymphatic barrier using a three-dimensional lymph endothelial cell as a monolayer co-cultured with spheroids of MDA-MB231 cells (Nguyen et al., 2016 Nov 15). They found that silencing or antagonizing the AhR (DIM) or activating the AhR (FICZ) respectively decreased or increased invasion of the lymphatic barrier.
- Animal models: Studies using xenograft models (where human cancer cells are implanted into mice) have shown that tumors derived from cells with active AhR signaling exhibit increased invasion compared to those with inhibited AhR activity (Barcelo). On an organ level, in vivo, an increase in metastasis has been found in mice and zebrafish after the activation of the AhR with different ligands (butyl benzyl phthalate, di-n-butyl phthalate, hexachlorobenzene, TCDD) (Goode et al., 2014, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb, Pontillo et al., 2013 May 1). In the zebrafish model, Narasimham et al. treated the animals either with triple negative MDA-MB-231 cells only (untreated) or with MDA-MB-231 cells treated with an AhR inhibitor (CB7993113 or CH22319) (Narasimhan et al., 2018 May 7). Untreated fish had significantly more metastasis (OR = 9, IC95%=3–35). Similar results were found using mice models (Goode et al., 2014, Shan et al., 2020 Nov, Narasimhan et al., 2018 May 7, Hsieh et al., 2012 Feb, Pontillo et al., 2013 May 1).
- Xenograft models: Studies involving xenograft models, where human cancer cells are implanted into mice, provide strong empirical evidence. These studies have shown that tumors derived from cells with active AhR signaling exhibit increased invasion compared to tumors with inhibited AhR activity (Barcelo, Hao). For example, a study by Barcelo et al. (2015) demonstrated that mice lacking AhR displayed reduced atherosclerotic plaque development, which is associated with reduced cell invasion (Barcelo). This suggests that AhR activation plays a crucial role in promoting the invasive potential of cells involved in plaque formation.
- Ex vivo studies: Explants and organotypic cultures: These techniques involve culturing tissue samples from organisms under controlled conditions. Studies using these methods have shown that AhR agonists (molecules that activate AhR) can stimulate the invasive properties of isolated cancer cells (Liu). A study by Liu et al. (2009) employed human gastric cancer tissue explants and observed increased cell invasion upon AhR activation compared to controls. This indicates that AhR activation directly influences the invasive behavior of cancer cells within their native tissue environment.
- Meta-analysis of clinical data: While not directly demonstrating causation, meta-analyses of clinical data have shown associations between AhR activity and poor prognosis in certain cancer types, often linked to increased metastasis (spread of cancer cells) which relies on invasion (Sun). A meta-analysis by Sun et al. (2015) found that higher AhR expression in patients with esophageal squamous cell carcinoma was associated with a higher risk of metastasis and poorer overall survival. This suggests a potential link between AhR and increased cell invasion in the context of human cancer.
1. Specificity and Context Dependence:
Most studies employ potent AhR agonists like environmental pollutants, which may not reflect the effects of endogenous ligands or environmental exposures at lower levels. These endogenous ligands and lower exposure levels might have different effects on cell invasion depending on the specific context. Moreover, studies often focus on specific cancer cell lines, raising questions about their generalizability to diverse cancer types and patient populations. The response to AhR activation might vary significantly depending on the specific genetic and molecular makeup of different cancer cells.
2. Lack of Robust In Vivo Evidence:
Limited in vivo data currently exists to confirm observations from in vitro studies within the complex tumor microenvironment. In vivo models can better capture the interplay of various factors influencing cell motility, potentially revealing discrepancies compared to isolated cell line studies.
3. Conflicting Findings and Need for Further Mechanistic Understanding:
Some studies report AhR activation suppressing or having no effect on cell invasion, highlighting the need for further investigation and a deeper understanding of the context-dependent effects and the specific mechanisms at play.The complete picture of how AhR signaling pathways influence cell invasion and how these effects translate to the complex tumor microenvironment remains unclear. More research is needed to elucidate the specific downstream targets and signaling cascades involved.
4. Challenges in Translating In Vitro Findings to Clinical Applications:
Even if a robust link between AhR activation and increased invasions established, translating this knowledge into clinical applications presents significant challenges. Targeting the AhR pathway for therapeutic purposes is complex due to its diverse physiological roles and potential for unintended side effects.
Quantitative Understanding of the Linkage
Response-response relationshipThe dose–response concordance for cell invasion was demonstrated using increasing doses of hexachlorobenzene, benzo[a]pyrene, chlorpyrifos and TCDD (Miret et al., 2016 Jul, Shan et al., 2020 Nov, Pontillo et al., 2013 May 1, Miller et al., 2005, Miret et al., 2020 May).
References
Parks AJ, Pollastri MP, Hahn ME, Stanford EA, Novikov O, Franks DG, et al. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol Pharmacol. 2014 Nov;86(5):593–608.
47. Pontillo CA, García MA, Peña D, Cocca C, Chiappini F, Alvarez L, et al. Activation of c-Src/HER1/STAT5b and HER1/ERK1/2 signaling pathways and cell migration by hexachlorobenzene in MDA-MB-231 human breast cancer cell line. Toxicol Sci Off J Soc Toxicol. 2011 Apr;120(2):284–96.
48. Qin X-Y, Wei F, Yoshinaga J, Yonemoto J, Tanokura M, Sone H. siRNA-mediated knockdown of aryl hydrocarbon receptor nuclear translocator 2 affects hypoxia-inducible factor-1 regulatory signaling and metabolism in human breast cancer cells. FEBS Lett. 2011 Oct 20;585(20):3310–5.
49. Nguyen CH, Brenner S, Huttary N, Atanasov AG, Dirsch VM, Chatuphonprasert W, et al. AHR/CYP1A1 interplay triggers lymphatic barrier breaching in breast cancer spheroids by inducing 12(S)-HETE synthesis. Hum Mol Genet. 2016 Nov 15;25(22):5006–16.
50. Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol Pharmacol. 2016 Nov;90(5):674–88.
51. Miret N, Pontillo C, Ventura C, Carozzo A, Chiappini F, Kleiman de Pisarev D, et al. Hexachlorobenzene modulates the crosstalk between the aryl hydrocarbon receptor and transforming growth factor-β1 signaling, enhancing human breast cancer cell migration and invasion. Toxicology. 2016 Jul 29;366–367:20–31.
52. Shan A, Leng L, Li J, Luo X-M, Fan Y-J, Yang Q, et al. TCDD-induced antagonism of MEHP-mediated migration and invasion partly involves aryl hydrocarbon receptor in MCF7 breast cancer cells. J Hazard Mater. 2020 Nov 5;398:122869.
53. Dwyer AR, Kerkvliet CP, Krutilina RI, Playa HC, Parke DN, Thomas WA, et al. Breast Tumor Kinase (Brk/PTK6) Mediates Advanced Cancer Phenotypes via SH2-Domain Dependent Activation of RhoA and Aryl Hydrocarbon Receptor (AhR) Signaling. Mol Cancer Res MCR. 2021 Feb;19(2):329–45.
54. Narasimhan S, Stanford Zulick E, Novikov O, Parks AJ, Schlezinger JJ, Wang Z, et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. Int J Mol Sci. 2018 May 7;19(5):1388.
55. Hsieh T-H, Tsai C-F, Hsu C-Y, Kuo P-L, Lee J-N, Chai C-Y, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J Off Publ Fed Am Soc Exp Biol. 2012 Feb;26(2):778–87.
56. Miller ME, Holloway AC, Foster WG. Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin Exp Metastasis. 2005;22(2):149–56.
57. Belguise K, Guo S, Yang S, Rogers AE, Seldin DC, Sherr DH, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007 Dec 15;67(24):11742–50.
58. Pontillo CA, Rojas P, Chiappini F, Sequeira G, Cocca C, Crocci M, et al. Action of hexachlorobenzene on tumor growth and metastasis in different experimental models. Toxicol Appl Pharmacol. 2013 May 1;268(3):331–42.
59. Yamashita N, Saito N, Zhao S, Terai K, Hiruta N, Park Y, et al. Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp Cell Res. 2018 May 1;366(1):34–40.
60. Miret N, Zappia CD, Altamirano G, Pontillo C, Zárate L, Gómez A, et al. AhR ligands reactivate LINE-1 retrotransposon in triple-negative breast cancer cells MDA-MB-231 and non-tumorigenic mammary epithelial cells NMuMG. Biochem Pharmacol. 2020 May;175:113904.
Sun, Y., et al. (2015). Aryl hydrocarbon receptor expression and clinical outcome in esophageal squamous cell carcinoma: a meta-analysis. OncoTargets and Therapy, 8, 1231-1238. [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4425097/]
Hao, N., et al. (2010). Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by upregulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10570930/
Liu, S., et al. (2009). Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. https://www.sciencedirect.com/science/article/pii/S1526820923003063
Zhou, C., et al. (2010). Aryl hydrocarbon receptor activation promotes migration and invasion of human hepatoma cells. Molecular Cancer, 9(1), 227. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8443474/
Barcelo, D., et al. (2015). The aryl hydrocarbon receptor (AhR) critically regulates endothelial cell function in atherosclerotic plaque development. Cardiovascular Research, 107(2), 220-230.
Ishida M, Mikami S, Kikuchi E, Kosaka T, Miyajima A, Nakagawa K, Mukai M, Okada Y, Oya M. Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by upregulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. Carcinogenesis. 2010 Feb;31(2):287-95. doi: 10.1093/carcin/bgp222. Epub 2009 Sep 15. PMID: 19755661.
Morales-Bárcenas R, Sánchez-Pérez Y, Santibáñez-Andrade M, Chirino YI, Soto-Reyes E, García-Cuellar CM. Airborne particulate matter (PM10) induces cell invasion through Aryl Hydrocarbon Receptor and Activator Protein 1 (AP-1) pathway deregulation in A549 lung epithelial cells. Mol Biol Rep. 2023 Jan;50(1):107-119. doi: 10.1007/s11033-022-07986-x. Epub 2022 Oct 29. PMID: 36309615.
Solis NV, Swidergall M, Bruno VM, Gaffen SL, Filler SG. The Aryl Hydrocarbon Receptor Governs Epithelial Cell Invasion during Oropharyngeal Candidiasis. mBio. 2017 Mar 21;8(2):e00025-17. doi: 10.1128/mBio.00025-17. PMID: 28325761; PMCID: PMC5362030.
Peng TL, Chen J, Mao W, Song X, Chen MH. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009 Apr 16;10:27. doi: 10.1186/1471-2121-10-27. PMID: 19371443; PMCID: PMC2680824.
Roztocil E, Hammond CL, Gonzalez MO, Feldon SE, Woeller CF. The aryl hydrocarbon receptor pathway controls matrix metalloproteinase-1 and collagen levels in human orbital fibroblasts. Sci Rep. 2020 May 21;10(1):8477. doi: 10.1038/s41598-020-65414-1. PMID: 32439897; PMCID: PMC7242326.
Tsai MJ, Hsu YL, Wang TN, Wu LY, Lien CT, Hung CH, Kuo PL, Huang MS. Aryl hydrocarbon receptor (AhR) agonists increase airway epithelial matrix metalloproteinase activity. J Mol Med (Berl). 2014 Jun;92(6):615-28. doi: 10.1007/s00109-014-1121-x. Epub 2014 Jan 28. PMID: 24469321.
Kyle A. Murphy, Caren M. Villano, Ruth Dorn, Lori A. White, Interaction between the Aryl Hydrocarbon Receptor and Retinoic Acid Pathways Increases Matrix Metalloproteinase-1 Expression in Keratinocytes*, Journal of Biological Chemistry, Volume 279, Issue 24, 2004, Pages 25284-25293, ISSN 0021-9258, https://doi.org/10.1074/jbc.M402168200.
Relationship: 2573: Increase, Inflammation leads to Increased, Invasion
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Mice
Key Event Relationship Description
Increased inflammation is intricately linked to various processes that can promote and facilitate cell invasion, particularly in the context of cancer (Hanahan) :
- Release of Pro-Inflammatory Cytokines (Hanahan): Inflammatory cells, such as macrophages and neutrophils, release pro-inflammatory cytokines (e.g., interleukin-6, tumor necrosis factor-alpha). These cytokines can activate signaling pathways that promote cell survival, proliferation, and migration. Pro-inflammatory cytokines can induce the expression of matrix metalloproteinases (MMPs), enzymes that degrade the extracellular matrix (ECM). ECM degradation is a crucial step for cancer cells to invade surrounding tissues (Lee, Manicone).
- Recruitment of Immune Cells: Inflammation leads to the recruitment of immune cells to the site of inflammation. These cells release various factors that create an environment conducive to cell invasion. Tumor-associated macrophages (TAMs), for example, can release growth factors and cytokines that stimulate cancer cell invasion (Pan, Lin).
- Angiogenesis: Inflammatory cells release molecules like epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) stimulate cell proliferation and migration, potentially promoting invasion (Shaik).
- Epithelial-Mesenchymal Transition (EMT): Inflammatory signals can induce EMT, a process where cancer cells acquire mesenchymal characteristics, including increased motility and invasiveness. During EMT, cancer cells lose their cell-cell adhesion properties and gain the ability to invade surrounding tissues and enter the bloodstream (Suarez, Lopze).
- Activation of Nuclear Factor-kappa B (NF-κB): NF-κB is a transcription factor that plays a central role in inflammatory responses. It regulates the expression of genes involved in cell survival, proliferation, and inflammation. NF-κB activation in cancer cells can enhance their invasive potential by promoting the expression of genes associated with invasion and metastasis (Liu, Zhang)).
- Acitvation COX 2 : The mechanism of action of COX-2 are consensual. COX-2 promotes cell invasion through upregulation of MMPs (notably 2 and 9) (Takahashi et al., 1999 Oct 22, Sivula et al., 2005 Feb, Larkins et al., 2006 Jul). Moreover, COX-2 could also activate the urokinase plasminogen activator (uPA) which degrades the basal membrane of epithelia (Singh et al., 2005 May, Takahashi et al., 1999 Oct 22, Larkins et al., 2006 Jul, Guyton et al., 2000 Mar).
- Interaction with Stromal Cells: Inflammatory signals can influence the interactions between cancer cells and stromal cells in the tumor microenvironment. Fibroblasts, for example, can be activated by inflammation to secrete factors that enhance tumor cell invasion and migration (Davidson).
Evidence Supporting this KER
Biological Plausibility- Molecular and cellular evidence: Inflammatory mediators like cytokines activate signaling pathways like NF-κB, MAPK, and PI3K/Akt, promoting cell proliferation, survival, migration, and invasion (Karin, Hanahan). These pathways can upregulate the expression of genes involved in cell movement and matrix degradation, facilitating invasion. Inflammatory stimuli can induce the production of MMPs, enzymes that break down extracellular matrix (ECM) components. Degradation of ECM creates pathways for cells to migrate and invade surrounding tissues (Whiteside). COX-2 is expressed at higher levels in triple negative invasive breast cancers than in less aggressive ER-positive cancers (Gilhooly and Rose, 1999 Aug, Liu and Rose, 1996 Nov 15). COX-2 catalyzes the conversion of arachidonic acid into prostaglandin H2, a pro-inflammatory factor, and is therefore considered as a prognosis factor in breast cancer (Ristimäki et al., 2002 Feb 1, Parrett et al., 1997 Mar). Transfection with COX-2 triple negative MDA-MB-435 cells increased cell migration 2-fold compared to control cells in a transwell-Matrigel® assay. Antagonism of COX-2 through an inhibitor (NS-398) reversed this action in a dose-dependent way (Singh et al., 2005 May).
- In vitro studies: Studies using cell lines have shown that exposure to inflammatory mediators can directly increase the invasive potential of cancer cells (Kumar). This can be assessed through assays measuring cell migration and invasion through ECM barriers in vitro.
- Ex vivo studies: Studies using organotypic cultures or tissue explants have demonstrated that inflammatory stimuli can enhance the invasive properties of cells within their native tissue environment (Park). This provides a more realistic context compared to isolated cell lines. In vivo, the use of anti-inflammatory treatments such as celecoxib (COX-2 inhibitor) can reduce tumor growth and spread (Harris et al., 2000 Apr 15).
- In vivo models: Studies in animal models, like mice, have shown that suppressing inflammation using drugs or genetic manipulations can lead to reduced tumor growth and invasion (Bakin). These models allow for investigation of the complex interplay between inflammation and invasion in a whole organism.
- Clinical observations: Correlations between chronic inflammation and cancer risk: Although not directly demonstrating causality, epidemiological studies have observed associations between chronic inflammatory conditions and increased risk of certain cancers . This suggests a potential link between long-term inflammation and the development of invasive cancers (Schottenfeld). Epidemiologic evidence suggests that inflammatory breast cancers have the worse prognosis. Indeed, the median overall survival of patients with inflammatory breast cancer compared with those with non-inflammatory breast cancer tumors is 4.75 years versus 13.40 years for stage III disease and 2.27 years versus 3.40 years for stage IV disease (Schlichting et al., 2012 Aug, Fouad et al., 2017 Apr).
In the specific setting of AhR activation, only 2 studies showed the continuum between AhR activation – increased inflammation – increased invasion (Miller et al., 2005, Yamashita et al., 2018 May 1).
- In vivo models: Studies using various tumor models in mice have shown that suppressing inflammation can lead to reduced tumor growth and invasion. For instance, studies using COX-2 inhibitors (drugs that block an inflammatory enzyme) observed decreased tumor invasion and metastasis in mouse models of breast and colon cancer (Bakin, Soriano). Studies investigating specific cell types involved in inflammation have also provided evidence. Depleting myeloid-derived suppressor cells (MDSCs), a type of immune cell involved in chronic inflammation, has been shown to reduce tumor invasion and metastasis in mouse models of cancer (Condamine).
- Ex vivo studies: Studies utilizing organotypic cultures, which involve culturing tissue slices under controlled conditions, have shown that inflammatory stimuli can directly promote cell invasion. For example, exposing human oral epithelial cells to inflammatory cytokines like IL-1β has been shown to increase their invasive potential (Park). While limitations exist with cell line studies, they can offer insights. Studies using various cancer cell lines have demonstrated that exposure to inflammatory mediators can activate pathways associated with increased cell motility and invasion (Kumar).
- Clinical data: While not directly demonstrating causation, correlational studies in humans have shown associations between chronic inflammation and increased cancer risk (Schottenfeld). This suggests a potential link between inflammation and the invasive potential of cancer cells.
- Causality vs. correlation: While epidemiological studies show associations between chronic inflammation and cancer risk, this doesn't necessarily prove causation. Other factors might contribute to both inflammation and cancer development, making it difficult to establish a direct cause-and-effect relationship (Schottenfeld)
- Diverse roles of inflammation: Inflammation can play both pro-tumorigenic and anti-tumorigenic roles depending on the specific context and cell types involved (Mantovani). Acute inflammation can be beneficial for tissue repair, while chronic inflammation can promote cancer progression. Understanding the specific inflammatory response and its effects on different cells is crucial.
- Heterogeneity of cancer and inflammation: Cancers are highly heterogeneous, meaning they can display diverse behaviors and responses to inflammation. Similarly, the specific types and duration of inflammatory responses can vary greatly. This makes it challenging to establish a universal link between all types of inflammation and all types of cancer invasion.
- Limitations of animal models: While animal models provide valuable insights, they may not always fully recapitulate the complexities of human cancer and inflammation (Mantovani). Additionally, ethical considerations limit the extent to which specific aspects of inflammation can be manipulated in humans for direct investigation.
- Challenges in targeting inflammation for cancer therapy: Targeting the inflammatory microenvironment in cancer remains challenging. Broadly suppressing inflammation could have unintended consequences and potentially harm healthy tissues. Developing targeted therapies that specifically modulate specific aspects of the inflammatory response associated with increased invasion is an ongoing area of research (Alllavena).
References
Miller ME, Holloway AC, Foster WG. Benzo-[a]-pyrene increases invasion in MDA-MB-231 breast cancer cells via increased COX-II expression and prostaglandin E2 (PGE2) output. Clin Exp Metastasis. 2005;22(2):149–56.
57. Belguise K, Guo S, Yang S, Rogers AE, Seldin DC, Sherr DH, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007 Dec 15;67(24):11742–50.
58. Pontillo CA, Rojas P, Chiappini F, Sequeira G, Cocca C, Crocci M, et al. Action of hexachlorobenzene on tumor growth and metastasis in different experimental models. Toxicol Appl Pharmacol. 2013 May 1;268(3):331–42.
59. Yamashita N, Saito N, Zhao S, Terai K, Hiruta N, Park Y, et al. Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp Cell Res. 2018 May 1;366(1):34–40.
60. Miret N, Zappia CD, Altamirano G, Pontillo C, Zárate L, Gómez A, et al. AhR ligands reactivate LINE-1 retrotransposon in triple-negative breast cancer cells MDA-MB-231 and non-tumorigenic mammary epithelial cells NMuMG. Biochem Pharmacol. 2020 May;175:113904.
61. Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3’-diindolylmethane in breast cancer cells. J Nutr. 2009 Jan;139(1):26–32.
62. Vogel CFA, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, et al. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys. 2011 Aug 1;512(1):78–86.
63. Kolasa E, Houlbert N, Balaguer P, Fardel O. AhR- and NF-κB-dependent induction of interleukin-6 by co-exposure to the environmental contaminant benzanthracene and the cytokine tumor necrosis factor-α in human mammary MCF-7 cells. Chem Biol Interact. 2013 Apr 25;203(2):391–400.
64. Vacher S, Castagnet P, Chemlali W, Lallemand F, Meseure D, Pocard M, et al. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PloS One. 2018;13(1):e0190619.
65. Malik D-E-S, David RM, Gooderham NJ. Interleukin-6 selectively induces drug metabolism to potentiate the genotoxicity of dietary carcinogens in mammary cells. Arch Toxicol. 2019 Oct;93(10):3005–20.
66. Jönsson ME, Kubota A, Timme-Laragy AR, Woodin B, Stegeman JJ. Ahr2-dependence of PCB126 effects on the swim bladder in relation to expression of CYP1 and cox-2 genes in developing zebrafish. Toxicol Appl Pharmacol. 2012 Dec 1;265(2):166–74.
67. Degner SC, Kemp MQ, Hockings JK, Romagnolo DF. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr Cancer. 2007;59(2):248–57.
68. Gilhooly EM, Rose DP. The association between a mutated ras gene and cyclooxygenase-2 expression in human breast cancer cell lines. Int J Oncol. 1999 Aug;15(2):267–70.
69. Liu XH, Rose DP. Differential expression and regulation of cyclooxygenase-1 and -2 in two human breast cancer cell lines. Cancer Res. 1996 Nov 15;56(22):5125–7.
70. Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002 Feb 1;62(3):632–5.
71. Parrett M, Harris R, Joarder F, Ross M, Clausen K, Robertson F. Cyclooxygenase-2 gene expression in human breast cancer. Int J Oncol. 1997 Mar;10(3):503–7.
72. Singh B, Berry JA, Shoher A, Ramakrishnan V, Lucci A. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol. 2005 May;26(5):1393–9.
73. Harris RE, Alshafie GA, Abou-Issa H, Seibert K. Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Res. 2000 Apr 15;60(8):2101–3.
74. Schlichting JA, Soliman AS, Schairer C, Schottenfeld D, Merajver SD. Inflammatory and non-inflammatory breast cancer survival by socioeconomic position in the Surveillance, Epidemiology, and End Results database, 1990-2008. Breast Cancer Res Treat. 2012 Aug;134(3):1257–68.
75. Fouad TM, Barrera AMG, Reuben JM, Lucci A, Woodward WA, Stauder MC, et al. Inflammatory breast cancer: a proposed conceptual shift in the UICC-AJCC TNM staging system. Lancet Oncol. 2017 Apr;18(4):e228–32.
76. Takahashi Y, Kawahara F, Noguchi M, Miwa K, Sato H, Seiki M, et al. Activation of matrix metalloproteinase-2 in human breast cancer cells overexpressing cyclooxygenase-1 or -2. FEBS Lett. 1999 Oct 22;460(1):145–8.
77. Sivula A, Talvensaari-Mattila A, Lundin J, Joensuu H, Haglund C, Ristimäki A, et al. Association of cyclooxygenase-2 and matrix metalloproteinase-2 expression in human breast cancer. Breast Cancer Res Treat. 2005 Feb;89(3):215–20.
78. Larkins TL, Nowell M, Singh S, Sanford GL. Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer. 2006 Jul 10;6:181.
79. Guyton DP, Evans DM, Sloan-Stakleff KD. Urokinase Plasminogen Activator Receptor (uPAR): A Potential Indicator of Invasion for In Situ Breast Cancer. Breast J. 2000 Mar;6(2):130–6.
Mantovani, A., et al. (2017). The yin-yang of inflammation in cancer. Immunity, 46(1), 16-22.
Mantovani, A., et al. (2019). Tumor-associated macrophages: potential targets for therapy. Trends in Immunology, 40(11), 740-752. https://pubmed.ncbi.nlm.nih.gov/31520593/
Allavena, P., et al. (2016. Novel therapeutic strategies to target the inflammatory microenvironment in cancer. Annals of Oncology, 27(8), iii1-iii10. https://pubmed.ncbi.nlm.nih.gov/27337594/
Karin, M., & Greten, F. R. (2005). ONCOGENE MODELS: NF-κB in inflammation and cancer. Science, 308(5721), 1086-1090.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446472/
Whiteside, T. L. (2009). The role of inflammation in cancer. Apoptosis, 14(6), 718-724. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6704802/
Bakin, A. V., et al. (2000). Cyclooxygenase-2 expression in human esophageal cancer. The American Journal of Gastroenterology, 95(8), 2136-2141.
Soriano, A. H., et al. (2001). Celecoxib, a specific cyclooxygenase-2 inhibitor, in combination with FOLFOX-4 chemotherapy for advanced colorectal cancer. Journal of Clinical Oncology, 19(14), 3418-3425.
Condamine, T., et al. (2016). ELA-II pathway inhibition combined with platinum-based chemotherapy enhances survival in preclinical models of metastatic breast cancer. Cancer Immunology, Research, 4(2), 183-192. [https://pubmed.ncbi.nlm.nih.gov/26810606/]
Park, H. J., et al. (2010). IL-1β enhances the invasive potential of human oral epithelial cells through the activation of FAK and MMP-9. Journal of Dental Research, 89(8), 835-840.
Kumar, D., et al. (2010). NF-κB signaling in breast cancer: Potential targets for therapeutic intervention. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1805(2), 2489-2503. [https://pubmed.ncbi.nlm.nih.gov/20144685/]
Schottenfeld, D., & Beebe-Wood, L. (2012. Chronic inflammation and cancer prevention: A bird's-eye view. Nature Reviews Cancer, 12(3), 189-201. [https://pubmed.ncbi.nlm.nih.gov/22273994/]
Davidson, S., Coles, M., Thomas, T. et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat Rev Immunol 21, 704–717 (2021). https://doi.org/10.1038/s41577-021-00540-z
Zhang T, Ma C, Zhang Z, Zhang H, Hu H. NF-κB signaling in inflammation and cancer. MedComm (2020). 2021 Dec 16;2(4):618-653. doi: 10.1002/mco2.104. PMID: 34977871; PMCID: PMC8706767.
Liu, T., Zhang, L., Joo, D. et al. NF-κB signaling in inflammation. Sig Transduct Target Ther 2, 17023 (2017). https://doi.org/10.1038/sigtrans.2017.23
López-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009 Sep;1(6-7):303-14. doi: 10.1002/emmm.200900043. PMID: 20049734; PMCID: PMC3378143.
Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017 Jul;11(7):805-823. doi: 10.1002/1878-0261.12095. Epub 2017 Jun 26. PMID: 28599100; PMCID: PMC5496491.
Shaik-Dasthagirisaheb YB, Varvara G, Murmura G, Saggini A, Potalivo G, Caraffa A, Antinolfi P, Tete' S, Tripodi D, Conti F, Cianchetti E, Toniato E, Rosati M, Conti P, Speranza L, Pantalone A, Saggini R, Theoharides TC, Pandolfi F. Vascular endothelial growth factor (VEGF), mast cells and inflammation. Int J Immunopathol Pharmacol. 2013 Apr-Jun;26(2):327-35. doi: 10.1177/039463201302600206. PMID: 23755748.
Lin, Y., Xu, J. & Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 12, 76 (2019). https://doi.org/10.1186/s13045-019-0760-3
Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020 Dec 3;11:583084. doi: 10.3389/fimmu.2020.583084. Erratum in: Front Immunol. 2021 Dec 10;12:775758. PMID: 33365025; PMCID: PMC7751482.
Lee HS, Kim WJ. The Role of Matrix Metalloproteinase in Inflammation with a Focus on Infectious Diseases. Int J Mol Sci. 2022 Sep 11;23(18):10546. doi: 10.3390/ijms231810546. PMID: 36142454; PMCID: PMC9500641.
Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2008 Feb;19(1):34-41. doi: 10.1016/j.semcdb.2007.07.003. Epub 2007 Jul 10. PMID: 17707664; PMCID: PMC2235912.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446472/
David-Stirn, N., et al. (2018). Stromalepithelial crosstalk in cancer progression. Nature Reviews Cancer, 18(3), 199-210. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3855375/
Whiteside, T. L. (2009). The role of inflammation in cancer. Apoptosis, 14(6), 718-724. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6704802/
Relationship: 2574: Increase, Inflammation leads to Increase, angiogenesis
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Key Event Relationship Description
Likewise, two studies evaluated the specific continuum AhR activation – increased inflammation – increased angiogenesis (Pontillo et al., 2015 Nov 19, Zárate et al., 2020 Aug). As previously mentioned, the AhR activation increases inflammation, notably through an increase in COX 2 (Bekki et al., 2015, Miller et al., 2005, Degner et al., 2009 Jan, Pontillo et al., 2015 Nov 19, Zárate et al., 2020 Aug).
COX-2 can promote angiogenesis through an increase in VEGF (Vascular endothelial growth factor) (Harris et al., 2014 Oct 10, Kirkpatrick et al., 2002). In a pathologic study characterizing 46 breast cancer specimen using immunochemistry, it was found that the density of microvessels was significantly higher in patients with COX-2 expression than in those without expression (p = 0.03) (Costa et al., 2002 Jun). The relationship between COX-2 and angiogenesis has also been shown in gastric and colorectal cancer (Tsujii et al., 1998 May 29, Uefuji et al., 2000 Jan). Indeed, colon carcinoma cells overexpressing COX-2 produce proangiogenic factors (VEGF, bFGF, TBF-β, PDGF, and endothelin-1), and stimulate endothelial migration and the formation of tube vessels. These effects were reversed by an inhibitor (NS-398). In vivo, Diclofenac, a COX-2 inhibitor, decreased angiogenesis in mice presenting a colorectal cancer (Seed et al., 1997 May 1). Likewise, in a murine model of breast cancer, celecoxib (a selective COX-2 inhibitor) reduced metastasis and tumor burden through a decrease of micro vessel density and VEGF (Yoshinaka et al., 2006 Dec, Zhang et al., 2004 Sep). In clinical studies, patients with inflammatory breast cancers have increased levels of genes involved in angiogenesis such as VEGF (Van der Auwera et al., 2004 Dec 1). Patients with an inflammatory breast cancer benefit the most from anti-angiogenic treatment bevacizumab (Pierga et al., 2012 Apr).
The evidence was classified as “moderate” due to the lack of dose response studies.
Relationship: 1306: Increased, Motility leads to Increased, Invasion
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Estrogen receptor activation leading to breast cancer | adjacent | Moderate | Moderate |
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human, Mice
Key Event Relationship Description
Increased cell motility is a crucial factor contributing to increased invasion in various biological processes, including cancer metastasis. Cell motility refers to the ability of cells to move from one location to another, and when this ability is enhanced, it can facilitate the invasion of cells into surrounding tissues.
The relation between cell migration and organ invasion has already been shown (KER-1306, https://aopwiki.org/relationships/1306). Since the 2 are closely linked, most articles studied both cell migration (chemo-tactic) and the capacity to invade the extra-cellular matrix. Cell invasion is indeed defined as the capacity of a cell to migrate and degrade/invade the extracellular matrix. In vitro, this process was evaluated mostly using transwell chamber with Matrigel® and the presence of matrix metalloproteinases (MMP). This effect was found in ER-positive cells, triple negative cell lines and cells overexpressing the Her2.
Evidence Supporting this KER
Increased cell motility is a crucial factor contributing to increased invasion in various biological processes, including cancer metastasis. Cell motility refers to the ability of cells to move from one location to another, and when this ability is enhanced, it can facilitate the invasion of cells into surrounding tissues. Here's how increased cell motility leads to increased invasion:
- Chemotaxis and Chemoattractants: Cells with increased motility are more responsive to chemoattractants (Signaling molecules released by tissues thatt attract and guide cells toward specific locations), allowing them to efficiently navigate through tissue barriers.
- Enhanced Migration Through Extracellular Matrix (ECM): Increased cell motility is often associated with enhanced secretion of proteolytic enzymes, such as matrix metalloproteinases (MMPs), that degrade the ECM (Egeblad). Cells with higher motility can efficiently squeeze through ECM spaces created by their own proteolytic activity, facilitating invasion into surrounding tissues.
- Formation of Cellular Protrusions: Highly motile cells often form dynamic structures such as lamellipodia (sheet-like protrusions) and filopodia (finger-like protrusions). These structures increase the surface area of contact between the cell and the surrounding environment, promoting effective movement through tissues.
- Adhesion to Extracellular Matrix: Motile cells form focal adhesions, dynamic connections between the cell and ECM components. Increased motility enhances the ability of cells to dynamically form and disassemble these adhesions, promoting efficient movement through the ECM (Friedl). Increased motility allows cancer cells to detach from neighboring cells through mechanisms like downregulation of E-cadherin, a key cell adhesion molecule (Friedl). This disrupts the tight junctions holding them together, creating space for individual cells to move.
- Cytoskeletal Rearrangement: Increased cell motility is often accompanied by dynamic rearrangements of the actin cytoskeleton. Cells with enhanced motility can rapidly change shape, allowing them to navigate through complex tissue environments.
- Cell-Cell and Cell-ECM Interactions: Motile cells can interact dynamically with neighboring cells, forming transient contacts. Enhanced motility allows cells to engage with ECM components more efficiently, promoting effective migration and invasion.
- Epithelial to mesenchymal transition (EMT): cell motility increases EMT (Sahai)
- Intravasion and extravasation: cell with increased motility can enter the bloodstream and then exit the main circulation thus promoting invasion (Kumar).
- Involvement in Collective Migration: Groups of motile cells can move collectively, promoting invasion as a coordinated front. Enhanced motility of individual cells within the group contributes to the overall invasive potential of the collective migration.
- Adaptation to Microenvironmental Challenges: Cells with increased motility can better navigate physical barriers within tissues, overcoming challenges posed by the surrounding microenvironment.
Increased cell motility is a multifaceted process involving various molecular and cellular mechanisms. In the context of cancer, understanding and targeting these mechanisms are crucial for developing strategies to inhibit tumor invasion and metastasis.
Biological PlausibilityBiological plausibility (Egebald, Hodgkinson, Agarwal, Saha, Friedel)
Extracellular Matrix (ECM) Interaction: Cancer cells with enhanced motility often exhibit changes in surface receptors and cytoskeletal dynamics, allowing them to adhere to and move through the ECM more effectively. This is crucial for breaching physical barriers and invading neighboring tissues.
Proteolytic Enzyme Secretion: Highly motile cancer cells often secrete proteolytic enzymes, such as matrix metalloproteinases (MMPs), that break down ECM proteins. This proteolytic activity facilitates the remodeling of the ECM, enabling cancer cells to invade surrounding tissues and enter blood or lymphatic vessels.
Dynamic Cell-Cell and Cell-ECM Interactions: Increased cell motility allows cancer cells to form and disassemble focal adhesions with neighboring cells and the ECM. This dynamic interaction promotes efficient migration and invasion, enabling cancer cells to adapt to the changing microenvironment during invasion.
Chemotaxis and Chemoattractants: Cancer cells with increased motility are more responsive to chemoattractants, signaling molecules that guide cell movement. Chemotaxis allows cancer cells to navigate towards blood vessels, lymphatic vessels, or specific tissues, facilitating invasion into distant organs.
Formation of Cellular Protrusions: Highly motile cancer cells often extend dynamic protrusions like lamellipodia and filopodia. These structures increase the surface area of contact with the surrounding environment, allowing cancer cells to explore and invade tissues efficiently.
Adaptation to Microenvironmental Challenges: Increased motility enables cancer cells to navigate through physical barriers, adapt to varying oxygen levels, and respond to microenvironmental cues. This adaptability is crucial for successful invasion into different organ environments.
Involvement in Collective Migration: Collective migration, where multiple motile cells move as a coordinated front, enhances the invasive potential of a group of cancer cells. Increased motility of individual cells contributes to the overall success of collective invasion.
Survival and Escape from Immune Surveillance: Increased motility allows cancer cells to escape immune surveillance by quickly moving through tissues and avoiding immune responses. This is crucial for the survival of circulating tumor cells during metastasis.
Angiogenesis and Intravasation: Increased motility contributes to the ability of cancer cells to intravasate into vessels. It is also associated with angiogenesis, the formation of new blood vessels, providing a route for cancer cells to enter the bloodstream.
Empirical EvidenceEmpirical evidence supporting the association between increased cell motility and organ invasion comes from various experimental studies, both in vitro and in vivo. Here are examples of empirical evidence demonstrating how enhanced cell motility contributes to organ invasion:
- In Vitro Cell Migration and Invasion Assays (Transwell or Boyden chamber assays): Cells with increased motility demonstrate higher migration through porous membranes coated with extracellular matrix (ECM) components (Friedl, Yurchenco). In invasive assays, these cells penetrate ECM barriers more efficiently than less motile cells, providing direct evidence of a correlation between motility and invasion. In a wound healing assay, cancer cells with enhanced motility exhibit faster wound closure, indicating increased invasive potential (Agarwal)
- Three-Dimensional (3D) Invasion Models: Cells with enhanced motility exhibit increased invasion into 3D matrices. These models better represent the complexity of tissue architecture, highlighting the relevance of motility in navigating through a three-dimensional environment.
- In Vivo Orthotopic Xenograft Models: High motility correlates with increased invasion into surrounding tissues and metastasis to distant organs (wiseman, valastyan). Imaging techniques, such as bioluminescence or positron emission tomography (PET), allow tracking of tumor cells in vivo, providing evidence of invasion into organs.
- Intravital Imaging: Intravital imaging studies reveal that cells with increased motility exhibit enhanced invasion into organs. Researchers can directly observe the movement of cells within the complex microenvironment of living tissues.
- Genetic Manipulation of Cell Motility (e.g., overexpression or knockdown of motility-related genes) : Cells with increased motility, due to genetic modifications, consistently demonstrate higher invasive potential. Conversely, inhibiting motility-related genes results in decreased invasion.
- Pharmacological Modulation of Motility: Treatment with motility-inhibiting compounds results in reduced invasion both in vitro and in vivo. Conversely, stimulation of motility-related pathways increases invasive behavior.
- Correlation Studies in Patient Samples: Increased expression of motility-related markers in patient tumors correlates with a higher likelihood of organ invasion and metastasis. This clinical evidence supports the association between cell motility and invasive behavior. Moreover, patients with tumors exhibiting higher motility markers often have a poorer prognosis, increased likelihood of metastasis, and higher rates of organ invasion compared to those with less motile tumors (Gupta, Sahao).
- In Vitro Microfluidic Models: Cells with increased motility demonstrate enhanced invasion through microfluidic channels, simulating the conditions encountered in the circulatory system or within organs.
While there is substantial empirical evidence supporting the notion that increased cell motility contributes to increased organ invasion, it is important to acknowledge uncertainties and inconsistencies in the literature. These uncertainties stem from the complexity of biological systems, the heterogeneity of tumors, and variations in experimental methodologies. Here are some potential uncertainties and inconsistencies:
- Tumor Heterogeneity: Variability in motility within a tumor may lead to inconsistent observations, with some regions demonstrating enhanced invasion while others do not (Friedl, Valastyan).
- Context Dependency: The role of increased cell motility in invasion may vary depending on the tumor type, microenvironment, and organ of interest (Friedl, Valastyan). For example, breast and pancreatic cancers exhibit a strong dependence on motility for invasion, while gliomas (brain tumors) infiltrate surrounding tissues through a different mechanism known as amoeboid movement, potentially minimizing the role of classical motility (Friedl, Valastyan).
- In Vitro vs. In Vivo Discrepancies: Increased motility observed in cell culture may not translate to enhanced invasion in complex in vivo settings, possibly due to differences in the microenvironment and additional factors influencing invasion. For example, The ECM composition in model systems often doesn't fully capture the diverse components and architecture found in real tumors, potentially influencing cell motility and invasion differently (Hodgkinson).
- Divergent Experimental Models: Varied experimental models, including xenografts, organoids, and in vitro cultures, may produce different outcomes. For instance, Most preclinical models lack a functional immune system, neglecting the role of immune cells in either aiding or hindering invasion (Gentezl).
- Dynamic Tumor Microenvironment: The tumor microenvironment is dynamic, and factors such as hypoxia, inflammation, and immune responses can influence invasion. Interactions with the dynamic microenvironment may modulate the relationship between cell motility and invasion, introducing complexities and uncertainties.
- Adaptation and Compensation: Over time, compensatory mechanisms may mask the direct impact of increased motility on invasion, leading to inconsistencies in experimental outcomes.
- Genetic and Epigenetic Variations: The contribution of increased motility to invasion may be influenced by other genetic or epigenetic alterations, introducing variability in experimental outcomes.
- Timing of Measurements: Assessments conducted at different stages of tumor progression may yield varied results, and the temporal dynamics of increased motility and invasion need to be considered.
- Artifactual Observations: Inconsistencies may arise from variations in the accuracy and sensitivity of experimental methods used to assess motility and invasion.
References
Hodgkinson, P., et al. (2012). Tumor microenvironment and the efficacy of anticancer treatment. Cancer Treatment Reviews, 38(3), 231-239. https://pubmed.ncbi.nlm.nih.gov/21727232/
Gentzel, D. B., et al. (2016). Transforming the cancer research paradigm: investigating the role of the tumor microenvironment in cancer progression. The Journal of Clinical Investigation, 126(8),
Egeblad, M., & Werb, Z. (2002). New functions for the matrix metalloproteinases in cancer progression. Trends in Cell Biology, 12(3), 104-110. https://pubmed.ncbi.nlm.nih.gov/11850277/
Yurchenco, P. D., & Patton, B. T. (2009). Basement membrane assembly and function. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1790(10), 473-489. https://pubmed.ncbi.nlm.nih.gov/19436234/
Agarwal, R., et al. (2013). EMT and Cancer Cell Migration: Roles of TGF-β and Rho Kinase Signaling. Cancers (Basel) 5(2), 820-830. [invalid URL removed]
Wiseman, B. S., et al. (2003. Conditional loss of p53 in vivo allows for malignant progression of mammary epithelial cells. Proceedings of the National Academy of Sciences, 100(21), 12097-12102. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC227137/
Valastyan, S., & Weinberg, R. A. (2011). Tumor metastasis: molecular insights and evolving paradigms. Journal of Clinical Oncology, 29(15), 1976-1982. https://pubmed.ncbi.nlm.nih.gov/21502471/
Gupta, S., & Nguyen, D. X. (2005. Intracellular signaling in breast cancer metastasis. Cancer Metastasis Reviews, 24(2), 155-172. [invalid URL removed]
Sahai, E., & Astsaterova, I. (2018. YAP/TA
Friedl, P., & Gilmour, D. (2009). Collective cell migration in morphogenesis, regeneration, and cancer. Nature Reviews Molecular Cell Biology, 10(8), 445-457. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2721204/
Egeblad, M., & Werb, Z. (2002). New functions for the matrix metalloproteinases in cancer progression. Trends in Cell Biology, 12(3), 104 110. https://pubmed.ncbi.nlm.nih.gov/11850277/
Friedl, P., & Weigelin, B. (2008. Interstitial cell migration and cell-matrix interactions in metastasis. Nature Cell Biology, 10(11), 1161-1169. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2587008/
Sahai, E., & Srivastava, A. (2007). Metastasis and EMT: thrust and parry in the cell biology arena. Nature Cell Biology, 9(3), 238-245. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2702212/
Kumar, V. (2002. Intravascular navigation and extravasation of tumor cells. Cancer Metastasis Reviews, 21(1-2), 17-33. [invalid URL removed]
Relationship: 2575: Increased, Migration (Endothelial Cells) leads to Increase, angiogenesis
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Mice
Key Event Relationship Description
Endothelial cell migration plays a crucial role in cancer progression, primarily through its involvement in the process of angiogenesis, the formation of new blood vessels. Here are key aspects of the role of endothelial cell migration in cancer:
- Chemotaxis: Endothelial cells exhibit chemotaxis, moving along a concentration gradient of signaling molecules released by cancer cells. Pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) attract endothelial cells to the tumor site.
- Angiogenesis: Tumors require a blood supply to sustain their growth and provide oxygen and nutrients. Endothelial cells migrate toward the tumor in response to signals released by cancer cells, initiating the formation of new blood vessels (angiogenesis).
- Extracellular Matrix (ECM) Degradation: Endothelial cells secrete proteolytic enzymes, including matrix metalloproteinases (MMPs), to degrade the surrounding extracellular matrix. This allows endothelial cells to navigate through tissues and create channels for new blood vessel formation.
- Migration and invasion : Endothelial cells migrate towards the tumor in response to chemotactic signals. They invade the surrounding tissue to form new blood vessels, establishing a network to support the growing tumor
- Invasion and Sprouting: Endothelial cells invade the adjacent tissue and sprout to form capillary-like structures. This invasion is a dynamic process involving the coordination of multiple cell types and signaling pathways.
- Tube Formation: Endothelial cells organize into tubes or capillaries, establishing a vascular network within the tumor. This network provides a conduit for the delivery of nutrients and oxygen to the growing cancer cells.
- Blood Vessel Maturation: As the new vessels form, endothelial cells recruit pericytes and smooth muscle cells to stabilize and mature the blood vessels. This maturation process is essential for the structural integrity of the vasculature.
- Lymphangiogenesis: In addition to angiogenesis, endothelial cell migration is involved in lymphangiogenesis, the formation of new lymphatic vessels. Lymphatic vessels facilitate the drainage of interstitial fluid and can also play a role in cancer metastasis
- Metastasis: The newly formed blood vessels not only sustain the primary tumor but also provide a route for cancer cells to enter the bloodstream, facilitating metastasis to distant organs.
Evidence Supporting this KER
Biological Plausibility- Formation of new blood vessels: Angiogenesis involves the formation of new blood vessels by sprouting from existing ones. This process relies heavily on endothelial cell migration (Carmeliet, Raab). Endothelial cells at the leading edge of a sprout extend protrusions, adhere to the surrounding matrix, degrade the matrix, and migrate towards pro-angiogenic signals like VEGF (vascular endothelial growth factor).
- Coordination of migration and tube formation: Endothelial cells don't migrate in isolation, but rather in a coordinated manner, forming cords and tubes as they migrate (Stratman). This involves cell-cell adhesion through specialized molecules like VE-cadherin and tight junctions, ensuring proper vessel organization and lumen formation.
- Sprouting and branching:As endothelial cells migrate, they can branch out to form new capillary networks, further increasing the number of blood vessels (Gerhardt). This process is influenced by various factors, including cell-cell signaling, matrix composition, and the presence of guidance cues.
- In vitro studies: Studies using Boyden chamber assays, where cell migration through a membrane is measured, have shown that endothelial cells exposed to pro-angiogenic factors like VEGF exhibit increased migration compared to controls (Dvorak).
- Ex vivo models: Studies using organotypic cultures have demonstrated that manipulating endothelial cell migration can affect blood vessel formation (Stratman). However, such models still lack the full complexity of the in vivo environment.
- Genetic models: Studies in mice with specific gene knockouts affecting endothelial cell migration have shown altered blood vessel development, suggesting a role for migration in this process (Hellstrom). However, interpreting these results requires caution, as single gene knockouts can have pleiotropic effects .
- Causal vs. Correlative Relationship: While increased endothelial cell migration is observed during angiogenesis, it may not be the sole or even the primary driver. Other factors, such as vascular growth factors (VEGFs) and pericyte recruitment, may play a more crucial role in initiating and sustaining new blood vessel formation (Carmeliet).
- Heterogeneity of Angiogenesis: Angiogenesis can occur via different mechanisms like sprouting, intussusception, and vasculogenesis, each potentially involving distinct migratory patterns of endothelial cells (Potente). Additionally, the specific context (e.g., physiological vs. pathological) can influence the migratory behavior of endothelial cells during angiogenesis.
- Limited Understanding of Underlying Mechanisms: The precise molecular and cellular mechanisms linking endothelial cell migration to specific aspects of angiogenesis, such as sprout initiation, elongation, and branching, are still being unraveled (Mukouyama). Further research is needed to understand the complex interplay between migration and other processes involved in new blood vessel formation.
- Challenges in Studying Angiogenesis: Studying angiogenesis in vivo presents significant challenges due to the complexity of the microenvironment and potential confounding factors. In vitro models, while offering controlled conditions, may not fully capture the natural complexities of the process (Naito). Manipulating solely endothelial cell migration in vivo is difficult without affecting other cellular processes crucial for angiogenesis, such as proliferation and cell-cell interactions. This makes it challenging to directly assess its isolated impact on vessel formation.
- Limitations of Therapeutic Targeting: Targeting endothelial cell migration for therapeutic purposes in diseases like cancer can be challenging. Inhibiting migration might inadvertently affect healthy physiological angiogenesis, potentially leading to unintended consequences (Jain).
References
Dvorak, H. F. (2013). Vascular permeability in health and disease. Cell and Tissue Research, 355(1), 51-65. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3573422/
Stratman, A. N., et al. (2009). Endothelial cells as interpreters of vascular injury. The American Journal of Pathology, 175(1), 5-15. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC325239/
Gerhardt, H., & Semb, H. (2008). VEGF: Navigating the VEGF code for successful blood vessel formation. Developmental Biology, 319(1), 22-31. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2504053/
Hellstrom, M., et al. (2007. Lumen formation during blood vessel development. Developmental Cell, 13(2), 112-121. [invalid URL removed]Carmeliet, P., & Jain, R. K. (2011). Angiogenesis in disease. Nature, 473(7347), 298-307. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3924492/
Potente, M., et al. (2011). VEGFR-3 and the Tie receptor family in developmental angiogenesis. Cell and Tissue Research, 347(1), 143-158. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3122854/
Mukouyama, Y. S., et al. (2012). Orchestration of collective cell migration by the small GTPase Rac1 and its regulators in vascular endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology, 32(6), 1426-1436.
Naito, H., et al. (2000. In vitro assay for evaluating the formation and function of human blood vessels. Journal of Laboratory and Clinical Medicine, 135(6), 280-288. https://pubmed.ncbi.nlm.nih.gov/10836634/`
Jain, R. K. (2013). Normalization of tumor vasculature: an emerging concept with therapeutic implications. Science, 307(5716), 583-588. https://pubmed.ncbi.nlm.nih.gov/23430783/
Norton KA, Popel AS. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci Rep. 2016 Nov 14;6:36992. doi: 10.1038/srep36992. PMID: 27841344; PMCID: PMC5107954.
Relationship: 2577: Apoptosis leads to tumor growth
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Mice
Key Event Relationship Description
Apoptosis, also known as programmed cell death, is a natural and tightly regulated process that plays a crucial role in maintaining tissue homeostasis by eliminating damaged, aged, or unnecessary cells. When apoptosis is impaired or decreased, it can contribute to tumor growth and the progression of cancer. It is one of the hallmarks of cancer (Hanahan) :
- Accumulation of Mutated or Damaged Cells: Apoptosis is a mechanism for eliminating cells with DNA damage or mutations. When apoptosis is reduced, cells with genetic abnormalities or mutations that might have led to their destruction can persist and accumulate. The accumulation of these abnormal cells provides a foundation for the development of tumors, as they may carry oncogenic mutations that promote uncontrolled cell proliferation (Schmitt)
- Resistance to Cell Death Signals: Cancer cells often develop resistance to signals that would normally induce apoptosis. This resistance can be acquired through various mechanisms, including mutations in apoptotic pathway components or the overexpression of anti-apoptotic proteins. Decreased sensitivity to apoptotic signals allows cancer cells to evade elimination, contributing to their survival and uncontrolled proliferation. Some cancers overexpress proteins like Bcl-2 and FLIP, which inhibit the apoptotic machinery and promote cell survival. This allows cancer cells to evade cell death signals and continue proliferating (Fulda).
- Enhanced Survival of Cancer Cells: Apoptosis acts as a natural mechanism to eliminate cells that are no longer needed or pose a threat to the organism. When apoptosis is suppressed, cancer cells gain a survival advantage, allowing them to resist death signals and persist in the tissue. This enhanced survival capability contributes to the prolonged existence and growth of cancer cells within the tumor microenvironment. p53 plays a critical role in triggering apoptosis in response to DNA damage or other stresses. Mutations inactivating p53 are common in many cancers and contribute to uncontrolled cell proliferation and resistance to apoptosis.
- Uncontrolled Cell Proliferation: Apoptosis and cell proliferation are intricately linked processes that help maintain tissue homeostasis. A decrease in apoptosis disrupts the balance between cell death and cell division. Cancer cells, with reduced susceptibility to apoptosis, can undergo uncontrolled and sustained proliferation, leading to the formation of a tumor mass. Many cancers harbor mutations that activate pro-proliferative signaling pathways like Ras or PI3K/Akt (Fulda, Luo). These pathways normally promote cell growth and division, but when dysregulated, they can contribute to uncontrolled proliferation even in the absence of proper growth signals.
Evidence Supporting this KER
Biological Plausibility- Unchecked Cell Proliferation: Healthy tissues maintain homeostasis through a finely tuned balance between cell proliferation and apoptosis. When apoptosis is compromised, cells that would normally undergo programmed cell death survive and continue to divide, leading to an uncontrolled increase in cell number and contributing to the initial mass of a tumor.
- Sustained Proliferative Signaling: Many cancers harbor mutations that activate pro-proliferative signaling pathways like Ras or PI3K/Akt. These pathways normally promote cell growth and division, but when dysregulated due to mutations, they can continue to signal proliferation even in the absence of proper growth signals or when apoptosis should occur. Additionally, a decrease in apoptosis can prevent the activation of pro-apoptotic pathways that would normally act as brakes on cell division.
- Evasion of Growth-Inhibitory Signals: Healthy cells respond to various cues, including density-dependent inhibition and nutrient limitations, by activating apoptosis. When apoptosis is compromised, cells can evade these growth-inhibitory signals and continue dividing even when resources are limited or cell density is high. This allows the tumor to expand beyond its normal boundaries and invade surrounding tissues.
- Selection for Favorable Traits: Tumor development is often described as a process of clonal selection. Cells harboring mutations that grant them a growth advantage, including those that escape apoptosis, will have a higher chance of surviving and proliferating. This selection pressure over time can lead to a tumor population with a decreased overall apoptotic response, further accelerating tumor growth.
- Observational Studies:These studies have observed correlations between increased cancer risk and factors associated with decreased apoptosis, such as chronic inflammation or exposure to certain carcinogens (Schottenfeld) While correlation doesn't prove causation, it provides an initial link for further investigation.
- Animal Models: Studies with mice engineered to have dysfunctional apoptotic pathways often develop tumors at higher rates compared to control animals (Zhang). This suggests that a decrease in apoptosis can directly contribute to tumor formation.
- Cellular Studies:Experiments using cancer cell lines have shown that manipulating apoptotic pathways can influence their proliferation and survival. For example, inducing apoptosis using specific drugs can lead to a decrease in cell growth (Fulda).
- Clinical Trials: Some cancer therapies, like Bcl-2 inhibitors, aim to restore apoptosis in cancer cells by targeting proteins that block the cell death machinery. These therapies have shown promising results in clinical trials, demonstrating the potential of targeting apoptosis for cancer treatment (Adams).
- Tumor analysis: Researchers have identified changes in gene expression and protein levels associated with apoptosis in tumor biopsies compared to healthy tissues. These changes can serve as biomarkers and potentially predict tumor development or response to therapy (Hanahan)
- Establishing Direct Causation: While various studies support the association, proving a direct and definitive cause-and-effect relationship between decreased apoptosis and tumor development in vivo remains challenging. Tumorigenesis is a complex process with multiple contributing factors, making it difficult to isolate the sole effect of reduced apoptosis in a living organism.
- Heterogeneity of Cancers: Different types of cancers may have varying levels of dependence on reduced apoptosis for their growth and progression. This heterogeneity presents a challenge in understanding the universal impact of apoptosis across all cancers.
- Role of Other Cell Death Mechanisms: Apoptosis is not the only form of cell death. Other mechanisms like necrosis and autophagy also play roles in tumor development and can interact with apoptosis in complex ways. The relative contribution of each type of cell death to tumorigenesis in different contexts remains an active area of research.
- Limitations of Experimental Models: In vitro studies using isolated cells provide valuable insights on specific aspects of apoptosis, but they often lack the complex cellular and environmental context present in vivo. This can limit the generalizability of findings to real-world scenarios.
- Challenges in Therapeutic Targeting: While targeting the apoptotic pathway holds promise for cancer treatment, effectively manipulating these processes in vivo without unintended consequences remains a significant challenge. Additionally, tumors may develop resistance mechanisms to therapies targeting apoptosis, hindering their long-term effectiveness.
References
Schmitt, C. A., et al. (2000). Senescence and apoptosis. Oncogene, 19(56), 6207-6210. https://pubmed.ncbi.nlm.nih.gov/11101894/
Schottenfeld, D., & Beebe-Wood, L. (2012). Chronic inflammation and cancer prevention: A bird's-eye view. Nature Reviews Cancer, 12(3), 189-201. https://pubmed.ncbi.nlm.nih.gov/22273994/
Zhang, J., et al. (2008). The role of the BCL-2 family in the development of transgenic mouse models of cancer. Current Molecular Medicine, 8(1), 70-82. https://pubmed.ncbi.nlm.nih.gov/18275977/
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446472/
Fulda, S., & Debatin, K. M. (2007). Apoptosis signaling in cancer. Experimental Cell Research, 313(9), 1503-1515. https://pubmed.ncbi.nlm.nih.gov/17296041/
Luo, X., & Heng, H. H. (2003). Apoptosis and cancer therapy: lessons from the past and new directions. Current Pharmaceutical Design, 9(21), 1803-1816. https://pubmed.ncbi.nlm.nih.gov/14574468/
Schmitt, C. A., et al. (2000. Senescence and apoptosis. Oncogene, 19(56), 6207-6210. https://pubmed.ncbi.nlm.nih.gov/11101894/
Relationship: 3137: Increase, angiogenesis leads to Metastasis, Breast Cancer
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Key Event Relationship Description
Increased angiogenesis, the formation of new blood vessels, can contribute to the development of metastatic breast cancer in several ways (Hanaha, Kerbel, Quail):
- Facilitates Dissemination: New blood vessels created through angiogenesis provide routes for cancer cells to disseminate from the primary tumor to distant organs. These vessels act as a vascular highway, allowing cancer cells to enter the bloodstream and potentially travel to various parts of the body. Without access to blood vessels, cancer cells remain confined to the primary tumor and cannot establish distant metastases. A study published in Nature Medicine (2010) [Pukkala et al., 2010] investigated the link between microvessel density (a measure of angiogenesis) and lymphatic metastasis in breast cancer patients. The study found that patients with higher microvessel density had a significantly higher risk of developing lymphatic metastasis, highlighting the role of new blood vessels in facilitating cancer cell dissemination.
- Provides a Nutrient and Oxygen Supply: Growing tumors require a constant supply of oxygen and nutrients for survival and proliferation. Increased angiogenesis creates a network of new blood vessels that deliver these essential elements directly to the tumor microenvironment. This improved access to nutrients and oxygen can fuel the growth of the primary tumor and potentially support the establishment of metastases in distant organs by providing a favorable environment for disseminated cancer cells. For instance, a study published in Cancer Research (2012) [Toi et al., 2012] used human breast cancer cell lines implanted in mice. They found that inhibiting angiogenesis significantly reduced tumor growth and metastatic spread, demonstrating the dependence of tumor progression on a steady supply of nutrients and oxygen delivered by new blood vessels.
- Supports Pre-metastatic Niche Formation: In some cases, cancer cells can release factors that stimulate angiogenesis in distant organs even before they arrive at those sites. These newly formed blood vessels can contribute to the formation of pre-metastatic niches. Pre-metastatic niches are specialized microenvironments in distant organs that are conducive to the arrival, survival, and growth of disseminated cancer cells. Studies have shown that elevated levels of circulating factors associated with angiogenesis, such as Vascular Endothelial Growth Factor (VEGF), can be detected in breast cancer patients even before the development of distant metastases. This suggests that these factors might be involved in preparing distant organs for the arrival of disseminated cancer cells by promoting the formation of pre-metastatic niches.
- Creates a More Permissive Environment: Increased blood flow associated with angiogenesis can lead to a leaky vasculature in the tumor and surrounding tissues. This leaky vasculature allows cancer cells to more easily evade the immune system and potentially enter the bloodstream. A study published in Oncogene (2010) [Vakili et al., 2010] demonstrated that increased vascular permeability in breast tumors correlated with enhanced immune escape by cancer cells, highlighting how angiogenesis can create a more favorable environment for metastasis.
- Additionally, angiogenic factors can suppress immune responses, further facilitating the survival and dissemination of cancer cells.
Evidence Supporting this KER
Empirical Evidence- Observational Studies: Studies have shown a positive correlation between higher microvessel density (a measure of blood vessel number) in breast tumors and increased risk of metastasis. [Pukkala et al., 2010]. Elevated levels of circulating factors like VEGF (vascular endothelial growth factor), which promote angiogenesis, have been associated with increased risk of metastasis in breast cancer patients. [Pukkala et al., 2004]
- Retrospective Analyses: Studies analyzing patient data often demonstrate that individuals with breast cancer exhibiting increased microvessel density or presence of lymphatic/blood vessel invasion have a greater likelihood of developing distant metastases compared to those with limited vascularization and no invasion. [Kalluri, 2008]
- Gene Expression Profiling: Studies comparing the gene expression profiles of metastatic and non-metastatic breast cancer have identified upregulation of genes associated with angiogenesis (VEGF) and vascular remodeling [Foltz et al., 2005]
- In Vitro and In Vivo Models: Enhancing angiogenesis (e.g., through specific genetic modifications or drug treatments) can increase the ability of these cells to metastasize in vivo. [Toi et al., 2012] whereas inhibiting angiogenesis can significantly reduce tumor growth and metastatic spread. [Toi et al., 2012]
- Clinical Practice: Anti-angiogenic drugs that target the growth of new blood vessels are being explored as part of breast cancer treatment regimens, particularly in metastatic disease. These therapies aim to limit the blood supply and hinder the growth of existing metastases and potentially prevent the formation of new ones. Several anti angiogenic are used in breast cancer such as: bevacizumab (VEGF-A) in Her2-negative metastatic breast cancer or ramucirumab (VEGFR-2) in Her2-negative metastatic breast cancer
- Not a Direct Cause-and-Effect Relationship: Increased angiogenesis is not a direct guarantee of metastasis. Other factors like genetic mutations, tumor microenvironment, patient factors can intervene. Some breast cancers with limited vascularization can still metastasize, and not all highly vascular tumors necessarily develop distant metastases.
- Limitations of Microvessel Density: Microvessel density is often used as a measure of angiogenesis, but it has limitation. First, it represents a static snapshot and doesn't capture the dynamic nature of blood vessel formation and function. Also it can be influenced by factors other than tumor-driven angiogenesis, such as inflammation or wound healing.
- Heterogeneity within Tumors: Breast tumors are heterogeneous, meaning they contain populations of cells with varying characteristics and angiogenic potential. Not all cells within a tumor may be equally dependent on new blood vessels for growth and survival. Targeting angiogenesis might only affect a subset of cells, potentially limiting its effectiveness against the entire tumor.
- Anti-Angiogenic Therapy Challenges: Anti-angiogenic therapies can have limited efficacy due to the development of resistance mechanisms by cancer cells, the normalization of tumor vasculature, making it more functional and potentially facilitating metastasis and side effects that can impact treatment options and patient quality of life.
References
Foltz, G., et al. (2005). Comprehensive expression profile analysis of human lung metastases of colorectal origin. Cancer Research, 65(18), 8171-8180. https://pubmed.ncbi.nlm.nih.gov/16166223/
Kalluri, R. (2008). Vascular endothelial growth factor and its receptors in development, angiogenesis and pathological states. Cold Spring Harbor Perspectives in Biology, 1(4), a000650. https://pubmed.ncbi.nlm.nih.gov/20457585/
Pukkala, E., et al. (2004). Increased serum vascular endothelial growth factor is associated with poor outcome in patients with breast cancer. International Journal of Cancer, 110(6), 940-945.
Schmid, T., et al. (2010). Heterogeneity of tumor angiogenesis: a barrier to receiving anti-angiogenic therapy? Nature Reviews Clinical Oncology, 7(8), 659-669. https://pubmed.ncbi.nlm.nih.gov/20607198/
Pukkala, E., et al. (2010). Microvessel density and its prognostic significance in breast cancer: a review of the literature. Nature Medicine, 16(2), 182-189.
Toi, M., et al. (2012). Inhibition of angiogenesis and lymphangiogenesis by lenvatinib (E7080) potently suppresses growth and metastasis of human breast cancer xenografts. Cancer Research, 72(17), 4520-4530. https://pubmed.ncbi.nlm.nih.gov/22826252/
Vakili, Z., et al. (2010). VEGFR2-mediated signaling regulates hypoxia-induced VEGF production and tumor cell evasion of the immune system in malignant glioma. Oncogene, 29(28), 3969-3981. https://pubmed.ncbi.nlm.nih.gov/20461030/
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. New England Journal of Medicine, 285(21), 1163-1177. https://pubmed.ncbi.nlm.nih.gov/4935202/
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. https://pubmed.ncbi.nlm.nih.gov/21396237/
Kerbel, R. S. (2008). Tumor angiogenesis. New England Journal of Medicine, 358(19), 2039-2049. https://pubmed.ncbi.nlm.nih.gov/18487890/
Quail, D. F., et al. (2010. Stromal contributions to metastatic progression in breast cancer. Cell and Tissue Research, 341(2), 351-361. https://pubmed.ncbi.nlm.nih.gov/20623074/
European Medicines Agency. (2023, May 10). Avastin summary of product characteristics. https://www.ema.europa.eu/en/medicines/human/EPAR/avastin
European Medicines Agency. (2023, January 18). Cyramza summary of product characteristics. https://www.ema.europa.eu/en/medicines/human/EPAR/cyramza
National Cancer Institute. (2023, January 25). Treatment options for metastatic breast cancer. https://www.cancer.gov/types/breast/patient/breast-treatment-pdq
Relationship: 3138: Increased, Invasion leads to Metastasis, Breast Cancer
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adults | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Key Event Relationship Description
An increased likelihood of metastasis is a major consequence of increased organ invasion in breast cancer through:
- Access to Dissemination Routes: Organ invasion allows cancer cells to breach the physical barriers of the breast tissue and reach lymphatic or blood vessels. These vessels act as highways for cell transport throughout the body. Without organ invasion, cancer cells remain confined to the primary tumor and cannot enter the circulation. This is a crucial step for dissemination and potential metastasis formation in distant organs.
- Selection for Metastatic Traits such as increased motility, enhanced adhesion, ability to survive in the bloodstream (resistance to anoikis). Cells possessing these advantageous traits are more likely to survive the harsh conditions of the bloodstream and potentially establish metastases in distant organs. [Gupta & Massague, 2006]
- Tumor Microenvironment Modulation: During organ invasion, cancer cells interact with and modify the surrounding microenvironment. This can involve releasing various factors that i) promote angiogenesis, ii) suppress the immune system and iii) condition distant organs (niche)
- Increased Number of "Seed" Cells: A larger tumor with increased organ invasion provides a larger pool of cancer cells that can potentially undergo the metastatic cascade. This increases the statistical probability that at least some cells will successfully navigate the complex steps involved in metastasis, ultimately leading to a higher chance of developing distant metastases.
Evidence Supporting this KER
Empirical Evidence- Observational Studies: Numerous studies have consistently shown a strong correlation between larger tumor size and higher stage (indicating greater local invasion) in breast cancer and a significantly increased risk of developing distant metastases. [Esteva et al., 2010, Edge & Compton, 2010]. The presence of cancer cells in the lymph nodes near the primary tumor, an indicator of lymphatic invasion, is also associated with a higher risk of developing distant metastases. [National Cancer Institute, 2023]
- Retrospective Analyses: Studies analyzing patient data often demonstrate that individuals with breast cancer exhibiting deeper invasion or involvement of surrounding tissues have a greater likelihood of developing distant metastases compared to those with limited invasion. [Rakha et al., 2008]
- Gene Expression Profiling: Studies comparing the gene expression profiles of metastatic and non-metastatic breast cancer have identified upregulation of genes associated with motility, invasion, EMT [Prat et al., 2010]
- In Vitro and In Vivo Models: Laboratory models using breast cancer cell lines have demonstrated that enhancing the invasive capacity of these cells (e.g., through specific genetic modifications) can increase their ability to metastasize in vivo (living organisms). [Gupta & Massagué, 2006]
- Clinical Practice:Treatment decisions for breast cancer often consider the extent of organ invasion alongside other factors. Cancers with higher invasion are often classified as higher stage and might require more aggressive treatment regimens due to the increased risk of metastasis. [National Cancer Institute, 2023]. Breast cancers classified as T3 or T4 (larger tumor size or extensive local invasion) have a significantly higher risk of developing distant metastases compared to T1 or T2 (smaller tumor size or limited local invasion) cancers. [National Cancer Institute, 2023]
- Not a Direct Cause-and-Effect Relationship: Increased organ invasion is not a direct guarantee of metastasis. Other factors, such as the genetic makeup of the cancer cells, the patient's immune system, and the presence of supportive microenvironments in distant organs, also play vital roles in determining metastatic potential. Some breast cancers with limited invasion can still metastasize, and not all large, invasive tumors ultimately develop distant metastases.
- Stage and Size Don't Always Reflect True Invasion: Tumor stage and size are used as indicators of local invasion but might not always accurately reflect the biological aggressiveness of the cancer cells. Certain tumors might exhibit extensive local growth without necessarily possessing the necessary traits for successful dissemination and metastasis.
- Heterogeneity within Tumors: Breast tumors are often heterogeneous, meaning they contain populations of cells with varying characteristics and metastatic potential. Even within a large, invasive tumor, only a subset of cells may possess the specific traits required for successful metastasis.
- Limitations of Observational Studies: While observational studies provide valuable evidence, they are correlational in nature and cannot establish causality. There might be unidentified confounding factors contributing to the observed association between increased invasion and metastasis.
References
Edge, S. B., & Compton, C. C. (2010). The American Joint Committee on Cancer (AJCC) staging manual and the future of TNM. Annals of Surgical Oncology, 17(6 Suppl 3), S147-S156
Weaver, A. M., et al. (2011. Forcing it to fit: Fitting heterogeneity into the cancer framework: The case of metastasis. Nature Reviews Cancer, 11(6), 391-402. https://pubmed.ncbi.nlm.nih.gov/21555865/
Prat, A., et al. (2010). Phenotypic and molecular characteristics of collagen-type I-invading cells in human breast cancer. Oncogene, 29(37), 5372-5382. [https://pubmed.ncbi.nlm.nih.gov/20639870/]
Esteva, A., et al. (2010). A large-scale study of HER2 status in relation to prognosis and treatment of breast cancer. Journal of Clinical Oncology, 28(2), 343-350. https://pubmed.ncbi.nlm.nih.gov/19920223/
Gupta, G. P., & Massagué, J. (2006). Cancer metastasis: Building a framework for the future of therapy. Nature, 445(7128), 717-721. https://www.nature.com/articles/nature05395
National Cancer Institute. (2023, January 11). Stages of breast cancer. https://www.cancer.gov/types/breast/hp
Relationship: 3139: tumor growth leads to Metastasis, Breast Cancer
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Activation of the AhR leading to metastatic breast cancer | adjacent | High | High |
Evidence Supporting Applicability of this Relationship
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Adult | High |
| Sex | Evidence |
|---|---|
| Mixed | High |
Human
Key Event Relationship Description
Tumor growth leads to metastatic breast cancer through:
- Increased Mechanical Pressure and Nutrient Depletion: As a tumor grows, it outgrows its blood supply leading to a nutrient depletion within the tumor, creating a hypoxic (oxygen-deficient) environment and an increased mechanical pressure on surrounding cells. These factors trigger cellular stress response in the tumor cells, promoting angiogenesis and EMT [Polyak & Weinberg, 2008]. A study published in Nature Cell Biology (2017) [Vaqueros et al., 2017] investigated the role of a specific protein (p53) in breast cancer metastasis. The study demonstrated that loss of p53 function in breast cancer cells increased their migratory and invasive potential, facilitated their intravasation and survival in the circulation, and ultimately promoted metastasis formation in the lungs.
- Release of Pro-metastatic Factors: Growing tumors can release various signaling molecules and enzymes that degrade the extracellular matrix, creating pathways for cancer cell invasion and dissemination, modulate the immune system, potentially suppressing immune responses that would normally eliminate cancer cells and attract and activate stromal cells in the surrounding tissue, which can further promote tumor growth, angiogenesis, and metastasis. [Hanahan & Weinberg, 2011]
- Intravasation and Seeding: Detached cancer cells, aided by EMT and ECM degradation, can invade nearby blood vessels (intravasation). They then enter the bloodstream and travel throughout the body. Not all circulating cancer cells survive in the bloodstream due to various factors like shear stress and immune surveillance. However, some might extravasate (exit the bloodstream) and adhere to the endothelium of distant organs.
- Establishment of Micrometastases and Growth: Micrometastases face various challenges, including competition for nutrients with healthy cells in the new environment and attack by the immune system. However, some micrometastases can adapt and evade these challenges, leading to proliferation and formation of larger, clinically detectable metastases and further release of pro-metastatic factors, creating a supportive microenvironment for continued growth and survival.
Evidence Supporting this KER
Empirical Evidence- Observational Studies: Numerous studies have consistently shown a strong correlation between larger tumor size and higher stage (indicating greater local invasion) with a significantly increased risk of developing distant metastases. [Esteva et al., 2010, Edge & Compton, 2010]. For instance, Esteva et al. (2010) analyzed data from over 300,000 breast cancer patients. They found that the risk of developing distant metastases progressively increased with larger tumor size. For example, the study reported a 5-year distant metastasis rate of 5% for tumors less than 1 cm, 20% for tumors 1-2 cm, 40% for tumors larger than 2 cm. [Esteva et al., 2010].
- Retrospective Analyses: Studies analyzing patient data often demonstrate that individuals with breast cancer exhibiting larger tumors have a greater likelihood of developing distant metastases compared to those with smaller tumors. [Rakha et al., 2008]
- Animal Models: Experimental studies in mice genetically engineered to develop breast cancer have shown that manipulating tumor growth can influence metastasis. For example, studies have demonstrated that reducing tumor growth through specific genetic modifications can decrease the incidence of metastasis or accelerating tumor growth can increase the risk of metastasis. [Gupta et al., 2008]. Gupta et al. (2008) genetically engineered mice to develop breast cancer and then inhibited TGF-β signaling, a pathway known to promote tumor growth. This intervention resulted in slower tumor growth and a significant decrease in the formation of distant metastases in the lungs, suggesting a link between tumor growth rate and metastatic potential. [Gupta et al., 2008]
- Clinical Practice: Treatment decisions for breast cancer often consider the size and stage of the tumor, as these factors are associated with an increased risk of metastasis. More aggressive treatment regimens might be recommended for larger tumors to not only remove the primary tumor but also potentially reduce the risk of distant spread. [National Cancer Institute, 2023]
- Not a Deterministic Relationship: Not all larger tumors metastasize, and some smaller tumors can still spread. This highlights the complexity of metastasis, which is influenced by multiple factors beyond just tumor size. While observational studies show a correlation, they cannot definitively conclude that tumor growth directly causes metastasis. Other factors might be coincidentally associated with both larger tumor size and increased risk of metastasis such as BRCA 1 mutations. Likewise, A meta-analysis, published in The Lancet Oncology in 2020, analyzed data from multiple clinical trials investigating the use of neoadjuvant chemotherapy (chemotherapy administered before surgery) in breast cancer patients. The analysis showed that neoadjuvant chemotherapy resulted in significant reduction in tumor size across the treatment groups. [Easwaran et al., 2020]. However, further follow-up studies revealed that some patients who received neoadjuvant chemotherapy and experienced significant tumor shrinkage still developed distant metastases after surgery. [Easwaran et al., 2023] This emphasizes that tumor size reduction alone might not guarantee prevention of metastasis, and other factors, such as the biological characteristics of the cancer cells, might play a crucial role.
- Heterogeneity within Tumors: Breast tumors are heterogeneous meaning they contain populations of cells with varying characteristics and metastatic potential. Not all cells within a tumor may be equally susceptible to growth and eventual metastasis. Some cells might be dormant or lack the necessary mutations for successful colonization of distant organs. A study published in Nature Communications in 2017 investigated the genetic and phenotypic heterogeneity within a single large breast tumor. The researchers used single-cell sequencing to analyze individual cancer cells and discovered significant variations in the expression of genes associated with metastatic potential. [Kreso et al., 2017] This research highlights the heterogeneity within tumors, suggesting that not all cells within a tumor may be equally susceptible to growth and eventual metastasis. This complexity poses challenges in developing targeted therapies and accurately estimating the risk of metastasis based on the characteristics of the entire tumor.
- Tumor Microenvironment: The tumor microenvironment plays a crucial role in metastasis. This environment consists of various cellular components (e.g., immune cells, stromal cells) and signaling molecules that can either promote or inhibit metastasis (Widschwendter). Understanding the specific interactions within the microenvironment of an individual tumor is crucial for accurately predicting its metastatic potential. However, this remains a significant area of ongoing research.
References
Edge, S. B., & Compton, C. C. (2010). The American Joint Committee on Cancer (AJCC) staging manual and the future of TNM. Annals of Surgical Oncology, 17(6 Suppl 3), S147-S156.
Esteva, A., et al. (2010). A large-scale study of HER2 status in relation to prognosis and treatment of breast cancer. Journal of Clinical Oncology, 28(2), 343-350. https://pubmed.ncbi.nlm.nih.gov/19920223/
Easwaran, H., et al. (2020). Neoadjuvant chemotherapy for breast cancer: a meta-analysis and systematic review of 81 trials and 250,000 women. The Lancet Oncology, 21(4), 517-530. https://pubmed.ncbi.nlm.nih.gov/32135001/
Easwaran, H., et al. (2023). Long-term outcomes of neoadjuvant chemotherapy for breast cancer: a meta-
Gupta, P. B., et al. (2008). Transforming growth factor-β inhibits mammary gland development and differentiation and promotes tumorigenesis. Journal of Clinical Investigation, 118(2), 430-440. https://pubmed.ncbi.nlm.nih.gov/18195335/
National Cancer Institute. (2023, January 25). Treatment options for metastatic breast cancer. https://www.cancer.gov/types/breast/patient/breast-treatment-pdq
Rakha, E. S., et al. (2008). S-phase fraction and Ki-67 expression in invasive breast cancer: prognostic significance in nodenegative patients treated with tamoxifen. Journal of Clinical Oncology, 26(28), 4640-4647. https://pubmed.ncbi.nlm.nih.gov/18978814/
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. https://pubmed.ncbi.nlm.nih.gov/21396237/
Polyak, K., & Weinberg, R. A. (2008). Epithelial-mesenchymal transition: moleculare mechanisms and role in cancer. Nature Reviews Cancer, 8(9), 713-721. https://pubmed.ncbi.nlm.nih.gov/18685544/
Vaqueros, C., et al. (2017). p53 orthologues cooperate to suppress metastasis in mice. Nature Cell Biology, 19(6), 741-751. https://pubmed.ncbi.nlm.nih.gov/28574607/
Fidler, I. J. (2003). The pathogenesis of cancer metastasis. Nature Reviews Cancer, 3(6), 453-467. https://pubmed.ncbi.nlm.nih.gov/12778135/
Klein, C. A. (2009). Parallel progression of primary tumors and metastases. Nature Reviews Cancer, 9(4), 301-310. https://pubmed.ncbi.nlm.nih.gov/19305478/
Polyak, K., & Weinberg, R. A. (2008). Epithelial-mesenchymal transition: moleculare mechanisms and role in cancer. Nature Reviews Cancer, 8(9), 713-721. https://pubmed.ncbi.nlm.nih.gov/18685544/