AOP ID and Title:
Graphical Representation
Status
| Author status | OECD status | OECD project | SAAOP status |
|---|---|---|---|
| Under development: Not open for comment. Do not cite |
Abstract
Decreased sperm count is a key endpoint in the assessment of male reproductive health as it is directly associated with impaired fertility. Exposure to DNA alkylating agents, including chemotherapeutic drugs and environmental toxicants, is associated with reduced sperm counts in experimental models and humans. However, the progression from DNA lesions to reduced sperm output has not been systematically organized in an AOP framework. This AOP addresses that gap by describing how DNA alkylation can lead to inadequate DNA repair, increased DNA strand breaks, apoptosis, impaired spermatogenesis, and ultimately decreased sperm count. Although genotoxicity data are not routinely used as predictors of male fertility effects, this AOP provides a basis for evaluating when such data may be informative for reproductive toxicity assessment and for developing future predictive toxicology approaches.
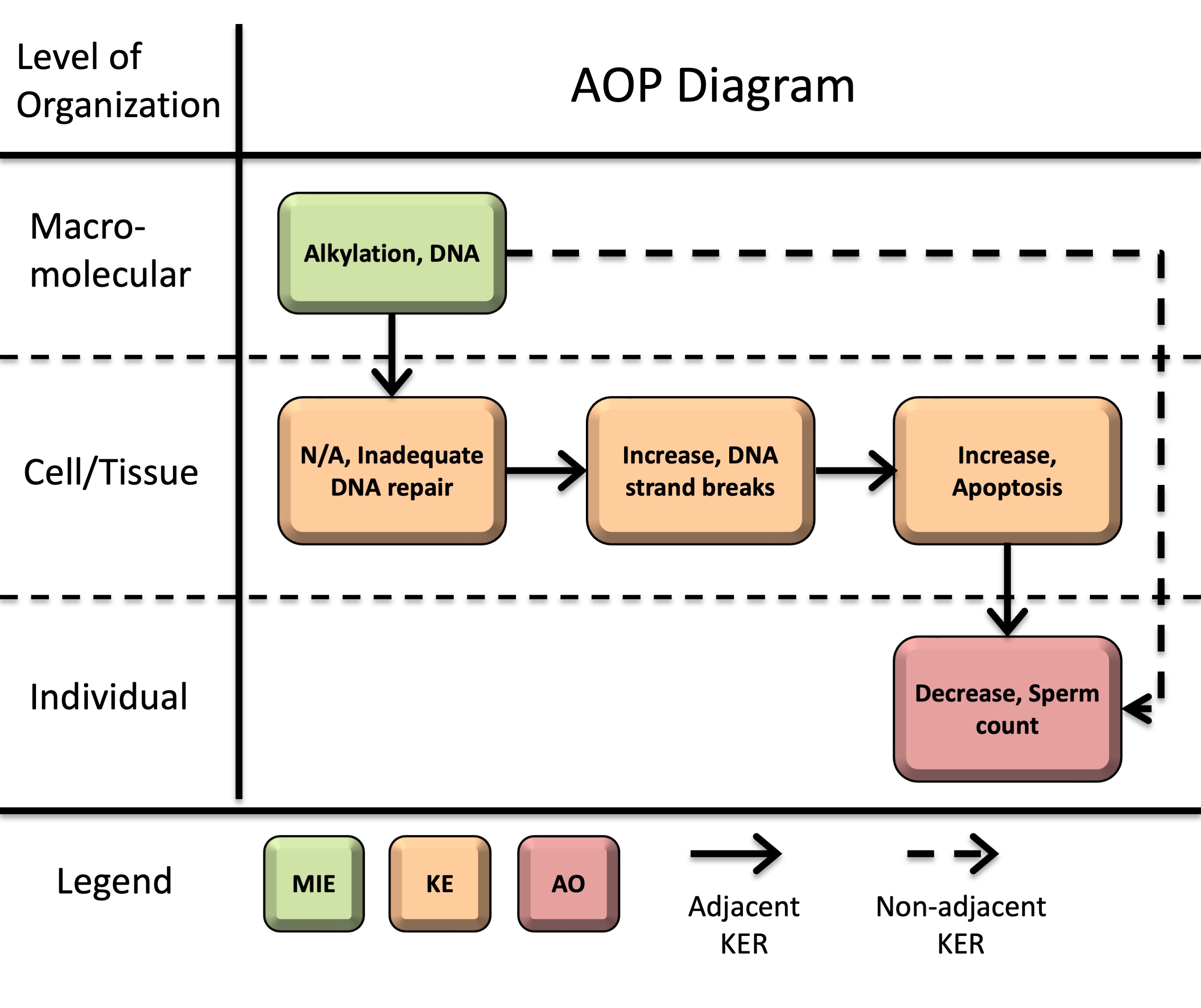
This AOP initiates with DNA alkylation (molecular initiating event, MIE). Alkylation-induced DNA lesions can then overwhelm DNA repair capacity (key event, KE1: inadequate repair) and an accumulation of DNA strand breaks (KE2). Persistent or unrepaired DNA damage activates DNA damage response pathways, ultimately leading to apoptosis (KE3). When apoptosis occurs in male germ cells and supporting testicular cells, excessive depletion of the developing germ cell population and disruption of structural support and endocrine signaling in the testis lead to decreased sperm counts (adverse outcome, AO) in sexually mature males.
This pathway is supported by strong biological plausibility and moderate to strong empirical evidence across multiple model systems, including human data, though quantitative understanding remains limited.
AOP Development Strategy
Context
The development of this AOP was motivated by the need to organize the well-established relationship between DNA alkylation and impaired male reproductive function into a formal mechanistic framework. DNA alkylation is a well-characterized form of genotoxic damage (Soll et al., 2017). In male spermatogonia and meiotic cells, alkylation of DNA in actively proliferating germ cells can trigger DNA damage responses, cell cycle arrest, and apoptosis, ultimately impairing spermatogenesis and leading to adverse reproductive outcomes (Kaina, 2003; Rübe et al., 2011; Li et al., 2025). Exposure to alkylating agents, particularly in the context of cancer chemotherapy, has long been associated with reduced sperm counts, oligozoospermia, azoospermia, and impaired fertility in males, with severity and recovery largely dependent on cumulative dose (reviewed by Howell and Shalet, 2005; Okada and Fujisawa, 2018). However, genotoxicity data are not routinely used as predictors of male fertility effects in reproductive toxicity assessment. This creates a need for a structured framework to evaluate when DNA damage in the male germline may be informative for reproductive hazard.
Human studies have reported associations between biomarkers of DNA alkylation and reduced sperm concentrations (Altakroni et al., 2021). Evidence from childhood cancer survivors also demonstrates that exposure during early life can impair germ cell populations and lead to reduced sperm production later in adulthood (Beaud et al., 2019; reviewed by Delessard et al., 2019). Experimental studies further demonstrate that alkylating agents produce dose-dependent and persistent reductions in sperm counts across species, including rodents, non-human primates, and humans (Meistrich, 1982a; Bucci and Meistrich, 1987; Hermann et al., 2009). Although DNA alkylation may occur during fetal, juvenile, or adult life stages, and can impair germ cell populations at any of these stages, the downstream events in this AOP involve spermatogenesis and sperm production; thus, the adverse outcome is manifested in sexually mature males.
This AOP branches from an existing AOP developed by Yauk et al., “Alkylation of DNA in Male Premeiotic Germ Cells Leading to Heritable Mutations” (AOP15), and contributes to the development of a broader AOP network for genotoxicity and reproductive toxicity.
An additional objective of this work is to facilitate the use of new approach methods (NAMs) in regulatory decision-making. By mechanistically linking early biological responses and adverse outcomes of regulatory concern, this AOP supports the development of novel models and screening tools to identify chemicals that may impair male fertility and provides a context for the use of data from NAMs. Additionally, by systematically organizing the existing knowledge on this topic we have identified key data gaps to guide future research in the field.
Summary of the AOP
Events
Molecular Initiating Events (MIE), Key Events (KE), Adverse Outcomes (AO)
| Sequence | Type | Event ID | Title | Short name |
|---|---|---|---|---|
| 1 | MIE | 97 | Alkylation, DNA | Alkylation, DNA |
| 2 | KE | 155 | Inadequate DNA repair | Inadequate DNA repair |
| 3 | KE | 1635 | Increase, DNA strand breaks | Increase, DNA strand breaks |
| KE | 1262 | Apoptosis | Apoptosis | |
| AO | 1757 | Decrease, Sperm count | Decrease, Sperm count |
Key Event Relationships
| Upstream Event | Relationship Type | Downstream Event | Evidence | Quantitative Understanding |
|---|---|---|---|---|
| Alkylation, DNA | adjacent | Inadequate DNA repair | High | Moderate |
| Inadequate DNA repair | adjacent | Increase, DNA strand breaks | High | Moderate |
| Increase, DNA strand breaks | adjacent | Apoptosis | High | Moderate |
| Apoptosis | adjacent | Decrease, Sperm count | High | Low |
| Alkylation, DNA | non-adjacent | Decrease, Sperm count | High | Low |
Overall Assessment of the AOP
Domain of Applicability
Life Stage Applicability| Life Stage | Evidence |
|---|---|
| Juvenile | High |
| Prepubertal | High |
| Adult, reproductively mature | High |
| Fetal | Moderate |
| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human | Homo sapiens | High | NCBI |
| Macaca mulatta | Macaca mulatta | Moderate | NCBI |
| rat | Rattus norvegicus | High | NCBI |
| mouse | Mus musculus | High | NCBI |
| Sex | Evidence |
|---|---|
| Male | High |
DNA alkylation can occur in all cell types. DNA repair processes and apoptotic pathways are conserved across species. While decreased sperm counts are measurable only after sexual maturity, the upstream KEs and KERs are biologically plausible and operative across fetal, juvenile, and adult life stages. Therefore, the overall biological domain of applicability of the AOP is considered relevant to male individuals exposed during fetal, juvenile, or adult stages, with manifestation of the AO occurring after reproductive maturation. In the male reproductive system, this AOP is most relevant when alkylation damage occurs in proliferating or meiotic germ cells that contribute directly to sperm production.
Essentiality of the Key Events
Overall, the essentiality of KEs in this AOP is supported primarily by indirect evidence from studies involving genetic manipulation, pharmacological intervention, and endpoint recovery, demonstrating that perturbation of upstream KEs is associated with corresponding changes in downstream KEs. A summary of the evidence supporting the essentiality of individual KEs and corresponding uncertainties or inconsistencies are provided in Table 1: summary of supporting evidence for essentiality of key events.
Impairment of DNA repair and DNA damage response (DDR) pathways consistently results in increased DNA strand breaks (KE2) and downstream apoptosis (KE3) in the presence of alkylating agents, indicating that upstream KEs are required for progression of the pathway. Although simultaneous measurements of alkyl DNA adducts, repair capacity, and sperm production within the same study are limited, exposure studies involving alkylating agents consistently show that greater DNA damage is associated with more severe and persistent impairment of sperm production across species, including rodents, non-human primates, and humans (Meistrich, 1982a, 1982b; Bucci and Meistrich, 1987; Hermann et al., 2009, Meistrich et al., 1992; Howell and Shalet, 2005; Okada and Fujisawa, 2018; Beaud et al., 2019). Together, these findings support the functional importance of DNA alkylation and DDR-related processes in progression of this AOP.
Essentiality of DNA strand breaks (KE2) is supported by studies showing that disruption of DDR signaling pathways (e.g., ATM inhibition) can prevent apoptosis even when DNA damage is present (Rodrigues et al., 2013). These findings highlight that damage sensing and downstream DDR signaling are required for the pathway to progress toward apoptotic cell death.
Evidence supporting the essentiality of apoptosis (KE3) is provided by intervention studies showing that attenuation of apoptotic signaling is associated with recovery of sperm counts (AO). Concordant reversibility across upstream and downstream endpoints following chemical or biological intervention supports the contribution of apoptosis to the AO (Oyovwi et al., 2023; Yaman et al., 2018; Udefa et al., 2020; Ehghaghi et al., 2022). However, many protective interventions also modulate oxidative stress and inflammatory pathways simultaneously, making it difficult to isolate apoptosis as the sole driver of sperm recovery.
Uncertainties include limitations in study design (e.g., reliance on single collection timepoints that do not capture temporal progression or delayed changes in sperm output), assay specificity (e.g., distinguishing primary DNA strand breaks from apoptotic DNA fragmentation), and incomplete characterization of quantitative relationships. Nonetheless, the overall weight of evidence supports a moderate level of essentiality.
Table 1. Summary of Supporting Evidence for Essentiality of Key Events
|
Event |
Direct Essentiality Evidence |
Indirect Essentiality Evidence |
Uncertainties or Inconsistency |
|
DNA alkylation (MIE) |
Limited direct evidence |
Exposure to alkylating agents leads to dose-dependent loss of germ cells and subsequent reductions in sperm counts (AO) across species; recovery may occur following removal of the stressor Repeated exposure to an alkylating agent caused a marked reduction in sperm counts in mice, with gradual recovery following cessation of exposure, demonstrating reversibility of the AO after removal of the stressor (Yin et al., 2014). |
Formation of alkyl DNA adducts in germ cells has been demonstrated in vivo; however, there are limited integrated measurements of the MIE, downstream KEs, the AO in the same studies. Consequently, progression through the pathway is often inferred from the known mechanisms of alkylating agents. |
|
Inadequate repair (KE1) |
Depletion of O6-alkylguanine-DNA alkyltransferase (AGT/MGMT) leads to corresponding alterations in DNA strand break (KE2) formation
Key studies: Roos et al. (2004) linked DNA alkylation (MIE), impaired repair (KE1), DNA strand breaks (KE2), and apoptosis (KE3) in proliferating human lymphocytes. Inactivation of MGMT increases persistence of alkylation-induced lesions, resulting in replication-dependent strand break formation and subsequent apoptosis.
Carlsson et al. (2025) showed that pharmacological inhibition of MGMT enhanced N-nitrosodimethylamine-induced formation of DNA adducts (MIE), DNA strand breaks (KE2), and micronucleus formation in HepG2-CYP2E1 human liver cells. |
Modulation of DNA repair (KE1) or DDR pathways leads to concurrent increases in DNA strand breaks (KE2) and apoptosis (KE3) Knockdown or knockout of CDKN2AIP, a regulator of DNA repair, leads to increased double strand breaks (DSBs) and apoptosis in mouse Sertoli cells and male germ cells (Cao et al., 2022).
Greater impairment of DDR pathways (Nbn/Atm double deletion) results in more DSBs and apoptotic cells than Nbn single deletion in mouse neuronal tissues (Rodrigues et al., 2013). Similarly, single or double deletion of Apc/p53 (Méniel et al., 2015), bidirectional genetic modulation of CIRKIL/Ku70 (Xiao et al., 2023), inhibition of the PI3K/mTOR pathway (Liu et al., 2014), or homologous recombination (Stringer et al., 2020), result in more DSBs and apoptosis than wildtype or single deletion models.
|
DNA repair capacity is not measured directly in many studies. Accumulation of DSBs following impairment of DDR pathways is interpreted as evidence of insufficient repair. |
|
DNA strand breaks (KE2) |
Blocking signaling downstream of DNA strand breaks (KE2) prevents apoptosis (KE3) |
Modulation of the magnitude of DNA strand breaks (KE2) is associated with a corresponding change in apoptosis (KE3) |
TUNEL staining may detect both primary strand breaks and apoptotic DNA fragmentation, when KE2 and KE3 are measured at overlapping timepoints. More specific DSB markers (e.g., γH2AX) were used in several studies.
|
|
Apoptosis (KE3) |
Limited direct evidence |
Attenuation of apoptosis (KE3) leads to recovery of sperm counts (AO) |
Protective agents often suppress inflammation and oxidative stress simultaneously. It is unclear if sperm recovery is due to reduced apoptosis or these co-activated pathways, or both. |
Weight of Evidence Summary
|
Biological plausibility of KERs |
Defining question |
High (Strong) |
Moderate |
Low (Weak) |
|
Is there a mechanistic relationship between KEup and KEdown consistent with established biological knowledge? |
Extensive understanding of the KER based on extensive previous documentation and broad acceptance. |
KER is plausible based on analogy to accepted biological relationships, but scientific understanding is incomplete. |
Empirical support for association between KEs, but the structural or functional relationship between them is not understood. |
|
|
MIE KE1: Alkylation, DNA leads to inadequate DNA repair |
STRONG Extensive evidence indicates that sufficiently high level of DNA alkylation can overwhelm cellular DNA repair machinery, leading to the persistence of DNA adducts and other unrepaired lesions. AGT, also known as MGMT in mammals, is an established suicide enzyme that can become saturated at high doses or after repeated exposure to alkylating agents, leading to inadequate DNA repair and accumulation of DNA alkyl adducts. This relationship is broadly conserved across species and cell types. |
|||
|
KE1 KE2: Inadequate DNA repair leads to Increase, DNA strand breaks |
STRONG DNA adducts and repair intermediates can accumulate when alkylation damage exceeds repair capacity in cells, including the depletion of AGT/MGMT. It is well established that persistence of unrepaired alkyl DNA lesions can interfere with DNA replication and promote replication fork stalling, leading to DNA strand breaks, which then activate DDR pathways. Extensive mechanistic evidence supports a causal relationship between inadequate DNA repair of alkylation-induced damage and increased DNA strand breaks. |
|||
|
KE2 KE3: Increase, DNA strand breaks leads to Apoptosis |
STRONG There is extensive mechanistic understanding of the DNA damage response pathways that link DNA strand breaks and apoptosis through both p53-dependent and independent mechanisms. |
|||
|
KE3 AO: Apoptosis leads to Decrease, Sperm Count |
STRONG Loss of testicular cells (e.g., developing germ cells and supportive somatic cells) through apoptosis disrupts normal testicular function to support spermatogenesis, resulting in a subsequent decrease in mature sperm output. These mechanisms are well established across mammalian systems. |
|||
|
MIE AO: Alkylation, DNA leads to Decrease, Sperm Count |
STRONG The mechanistic linkage is conserved across species and supported by extensive knowledge of germ cell biology and toxicology. While alkylation damage can occur across all stages of spermatogenesis, effects on sperm counts are primarily driven by damage to proliferating and meiotic germ cells, whereas damage to post-meiotic cells predominantly affects sperm quality rather than quantity. |
|||
|
Essentiality of KEs |
Defining question |
High (Strong) |
Moderate |
Low (Weak) |
|
Are downstream KEs and/or the AO prevented if an upstream KE is blocked? |
Direct evidence from specifically designed experimental studies illustrating essentiality for at least one of the important KEs. |
Indirect evidence that sufficient modification of an expected modulating factor attenuates or augments a KE. |
No or contradictory experimental evidence of the essentiality of any of the KEs. |
|
|
AOP-level |
MODERATE Evidence supporting the essentiality of KEs is available from genetic and mechanistic studies. Modulation of DNA damage response and apoptotic pathways induce corresponding changes in downstream outcomes, including apoptosis and sperm counts. A limited number of studies provide more direct evidence of essentiality for specific KERs, while identifying a few essential signaling mediators involved in the transduction of DNA damage into apoptosis. However, such direct evidence is not consistently available across all KEs in the pathway, and much of the support remains indirect or context-specific. |
|||
|
Empirical support for KERs |
Defining question |
High (Strong) |
Moderate |
Low (Weak) |
|
Does empirical evidence support that a change in KEup leads to an appropriate change in KEdown? |
Multiple studies showing dependent change in both events following exposure to a wide range of specific stressors. |
Demonstrated dependent change in both events following exposure to a small number of stressors. |
Limited or no studies reporting dependent change in both events following exposure to a specific stressor; and/or significant inconsistencies in empirical support across taxa and species that don’t align with hypothesized AOP. |
|
|
MIE KE1: Alkylation, DNA leads to inadequate DNA repair |
STRONG Inadequate DNA repair is measured indirectly through persistence of DNA adducts or increases in mutations that result from unrepaired DNA damage. Extensive evidence from somatic and germ cells supports this KER (in particular for temporal concordance), although quantitative dose-response concordance is less well characterized. There are no apparent inconsistencies. Numerous studies demonstrate that alkyl DNA adducts persist when repair capacity is exceeded or repair pathways are impaired. In particular, saturation or depletion of AGT/MGMT results in increased persistence of O6-alkylguanine adducts, providing strong support for this KER. |
|||
|
KE1 KE2: Inadequate DNA repair leads to Increase, DNA strand breaks |
MODERATE Limited in vivo data are available. However, multiple in vitro and genetic studies demonstrate that impairment of DNA repair or DDR pathways result in increased accumulation and persistence of DNA strand breaks in both somatic cells and germ cells following exposure to genotoxic stressors. In the context of DNA alkylation, saturation or depletion of AGT/MGMT leads to persistence of O6-alkylguanine lesions, which are subsequently converted into DNA strand breaks, consistent with temporal concordance. Studies involving AGT depletion and repair-deficient systems provide empirical support for the essential role of inadequate repair of alkylation DNA damage in the accumulation of DNA strand breaks. |
|||
|
KE2 KE3: Increase, DNA strand breaks leads to Apoptosis |
STRONG Temporal concordance is consistently observed, with DNA strand breaks occurring earlier or concurrently with apoptotic responses across in vitro somatic and germ cells, and rodent models. Dose concordance is supported, although dose-response data are limited in some studies. Evidence for incidence concordance is supported by a small number of studies, while others are limited by lack of appropriate measurements. |
|||
|
KE3 AO: Apoptosis leads to Reduce, Sperm Count |
STRONG Concordant changes between increased apoptosis and decreased sperm counts are have been consistently observed across multiple in vivo rodent studies. Temporal alignment is biologically supported, although it is often inferred rather than directly measured. Evidence for dose concordance is limited as many studies used a single exposure dose, preventing assessment of dose-dependent changes. Incidence concordance is generally not assessed, as both apoptosis and sperm count are typically reported as continuous outcomes (e.g., group means) rather than as the proportion of individual animals meeting predefined criteria for increased apoptosis or reduced sperm counts. Nevertheless, the consistency of the empirical evidence, together with multiple intervention studies demonstrating recovery of sperm counts following attenuation of apoptotic signaling, provides strong support for this KER. |
|||
|
MIE AO: Alkylation, DNA leads to Decrease, Sperm Count |
STRONG Empirical evidence from both experimental animal models and human studies supports a consistent relationship between DNA alkylation and reduced sperm counts. Although direct measurement of both KEs in the same study is limited, extrapolation across studies involving exposure to well-characterized alkylating agents provides strong empirical support for temporal and dose concordance. Reduction in sperm counts occur after delays consistent with spermatogenic progression, and higher exposures lead to greater and more sustained decreases in sperm counts across multiple studies and stressors. |
|||
Quantitative Consideration
The overall quantitative understanding of the KERs in this AOP is low. While individual KERs are supported by qualitative evidence of dose-response and temporal concordance, quantitative relationships between KEs are not well defined.
Some studies demonstrate graded changes between adjacent KEs following genetic or pharmacological modulation (e.g., KER2 and KER3), supporting response-response relationships. More details are provided in the individual KERs.
A threshold-based response is expected in this AOP, as DNA damage must exceed the repair capacity to propagate to downstream effects. In addition, a sufficient level of germ cell apoptosis is likely required before a measurable decline in sperm count occurs. However, several modulating factors have been identified, and the quantitative relationships are not generalizable across cell types, tissues, or developmental stages.
References
Altakroni, B., Nevin, C., Carroll, M., Murgatroyd, C., Horne, G., Brison, D. R. & Povey, A. C. (2021). The marker of alkyl DNA base damage, N7-methylguanine, is associated with semen quality in men. Scientific Reports, 11(1), 3121. https://doi.org/10.1038/s41598-021-81674-x
Beaud, H., Albert, O., Robaire, B., Rousseau, M. C., Chan, P. T. K. & Delbes, G. (2019). Sperm DNA integrity in adult survivors of paediatric leukemia and lymphoma: A pilot study on the impact of age and type of treatment. PLoS ONE, 14(12), e0226262. https://doi.org/10.1371/journal.pone.0226262
Bucci, L. R. & Meistrich, M. L. (1987). Effects of busulfan on murine spermatogenesis: cytotoxicity, sterility, sperm abnormalities, and dominant lethal mutations. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 176(2), 259–268. https://doi.org/10.1016/0027-5107(87)90057-1
Carlsson, M. J., Herzog, N., Felske, C., Ackermann, G., Regier, A., Wittmann, S., Cereijo, R. F., Sturla, S. J., Küpper, J.-H. & Fahrer, J. (2025). The DNA Repair Protein MGMT Protects against the Genotoxicity of N‑Nitrosodimethylamine, but Not N‑Nitrosodiethanolamine and N‑Nitrosomethylaniline, in Human HepG2 Liver Cells with CYP2E1 Expression. Chemical Research in Toxicology, 38(6), 1134–1146. https://doi.org/10.1021/acs.chemrestox.5c00133
Cao, Y., Sun, Q., Chen, Z., Lu, J., Geng, T., Ma, L. & Zhang, Y. (2022). CDKN2AIP is critical for spermiogenesis and germ cell development. Cell & Bioscience, 12(1), 136. https://doi.org/10.1186/s13578-022-00861-z
Delessard, M., Saulnier, J., Rives, A., Dumont, L., Rondanino, C. & Rives, N. (2020). Exposure to Chemotherapy During Childhood or Adulthood and Consequences on Spermatogenesis and Male Fertility. International Journal of Molecular Sciences, 21(4), 1454. https://doi.org/10.3390/ijms21041454
Ehghaghi, A., Zokaei, E., Modarressi, M. H., Tavoosidana, G., Ghafouri-Fard, S., Khanali, F., Motevaseli, E. & Noroozi, Z. (2022). Antioxidant and anti-apoptotic effects of selenium nanoparticles and Lactobacillus casei on mice testis after X-ray. Andrologia, 54(11), e14591. https://doi.org/10.1111/and.14591
Gur, C., Akarsu, S. A., Akaras, N., Tuncer, S. C. & Kandemir, F. M. (2023). Carvacrol reduces abnormal and dead sperm counts by attenuating sodium arsenite-induced oxidative stress, inflammation, apoptosis, and autophagy in the testicular tissues of rats. Environmental Toxicology, 38(6), 1265–1276. https://doi.org/10.1002/tox.23762
Hermann, B. P., Sukhwani, M., Lin, C., Sheng, Y., Tomko, J., Rodriguez, M., Shuttleworth, J. J., McFarland, D., Hobbs, R. M., Pandolfi, P. P., Schatten, G. P. & Orwig, K. E. (2009). Characterization, Cryopreservation, and Ablation of Spermatogonial Stem Cells in Adult Rhesus Macaques. Stem Cells, 25(9), 2330–2338. https://doi.org/10.1634/stemcells.2007-0143
Howell, S. J. & Shalet, S. M. (2005). Spermatogenesis After Cancer Treatment: Damage and Recovery. JNCI Monographs, 2005(34), 12–17. https://doi.org/10.1093/jncimonographs/lgi003
Kaina, B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochemical Pharmacology, 66(8), 1547–1554. https://doi.org/10.1016/s0006-2952(03)00510-0
Li, N., Wang, H., zou, S., Yu, X. & Li, J. (2025). Perspective in the Mechanisms for Repairing Sperm DNA Damage. Reproductive Sciences, 32(1), 41–51. https://doi.org/10.1007/s43032-024-01714-5
Liu, W.-L., Gao, M., Tzen, K.-Y., Tsai, C.-L., Hsu, F.-M., Cheng, A.-L. & Cheng, J. C.-H. (2014). Targeting Phosphatidylinositide3-Kinase/Akt pathway by BKM120 for radiosensitization in hepatocellular carcinoma. Oncotarget, 5(11), 3662–3672. https://doi.org/10.18632/oncotarget.1978
Meistrich, M. L. (1982a). Quantitative Correlation Between Testicular Stem Cell Survival, Sperm Production, and Fertility in the Mouse After Treatment With Different Cytotoxic Agents. Journal of Andrology, 3(1), 58–68. https://doi.org/10.1002/j.1939-4640.1982.tb00646.x
Meistrich, M. L., Finch, M., Cunha, M. F. da, Hacker, U. & Au, W. W. (1982b). Damaging effects of fourteen chemotherapeutic drugs on mouse testis cells. Cancer Research, 42(1), 122–131.
Meistrich, M. L., Wilson, G., Brown, B. W., Cunha, M. F. da & Lipshultz, L. I. (1992). Impact of cyclophosphamide on long-term reduction in sperm count in men treated with combination chemotherapy for Ewing and soft tissue sarcomas. Cancer, 70(11), 2703–2712. https://doi.org/10.1002/1097-0142(19921201)70:11<2703::aid-cncr2820701123>3.0.co;2-x
Méniel, V., Megges, M., Young, M. A., Cole, A., Sansom, O. J., Clarke, A. R. (2015). Apc and p53 interaction in DNA damage and genomic instability in hepatocytes. Oncogene, 34(31), 4118–4129. https://doi.org/10.1038/onc.2014.342
Murphy, C. J. & Richburg, J. H. (2015). Implications of Sertoli cell induced germ cell apoptosis to testicular pathology. Spermatogenesis, 4(2), e979110. https://doi.org/10.4161/21565562.2014.979110
Okada, K. & Fujisawa, M. (2018). Recovery of Spermatogenesis Following Cancer Treatment with Cytotoxic Chemotherapy and Radiotherapy. The World Journal of Men’s Health, 36(2), 166–174. https://doi.org/10.5534/wjmh.180043
Oyovwi, M. O., Oghenetega, O. B., Victor, E., Faith, F. Y. & Uchechukwu, J. G. (2023). Quercetin protects against levetiracetam induced gonadotoxicity in rats. Toxicology, 491, 153518. https://doi.org/10.1016/j.tox.2023.153518
Roos, W., Baumgartner, M. & Kaina, B. (2004). Apoptosis triggered by DNA damage O6-methylguanine in human lymphocytes requires DNA replication and is mediated by p53 and Fas/CD95/Apo-1. Oncogene, 23(2), 359–367. https://doi.org/10.1038/sj.onc.1207080
Rodrigues, P. M. G., Grigaravicius, P., Remus, M., Cavalheiro, G. R., Gomes, A. L., Rocha-Martins, M., Martins, M. R., Frappart, L., Reuss, D., McKinnon, P. J., Deimling, A. von, Martins, R. A. P. & Frappart, P.-O. (2013). Nbn and Atm Cooperate in a Tissue and Developmental Stage-Specific Manner to Prevent Double Strand Breaks and Apoptosis in Developing Brain and Eye. PLoS ONE, 8(7), e69209. https://doi.org/10.1371/journal.pone.0069209
Rübe, C. E., Zhang, S., Miebach, N., Fricke, A. & Rübe, C. (2011). Protecting the heritable genome: DNA damage response mechanisms in spermatogonial stem cells. DNA Repair, 10(2), 159–168. https://doi.org/10.1016/j.dnarep.2010.10.007
Soll, J. M., Sobol, R. W. & Mosammaparast, N. (2017). Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends in Biochemical Sciences, 42(3), 206–218. https://doi.org/10.1016/j.tibs.2016.10.001
Udefa, A. L., Amama, E. A., Archibong, E. A., Nwangwa, J. N., Adama, S., Inyang, V. U., Inyaka, G. U., Aju, G. J., Okpa, S. & Inah, I. O. (2020). Antioxidant, anti-inflammatory and anti-apoptotic effects of hydro-ethanolic extract of Cyperus esculentus L. (tigernut) on lead acetate-induced testicular dysfunction in Wistar rats. Biomedicine & Pharmacotherapy, 129, 110491. https://doi.org/10.1016/j.biopha.2020.110491
Xiao, H., Zhang, M., Wu, H., Wu, J., Hu, X., Pei, X., Li, D., Zhao, L., Hua, Q., Meng, B., Zhang, X., Peng, L., Cheng, X., Li, Z., Yang, W., Zhang, Q., Zhang, Y., Lu, Y. & Pan, Z. (2022). CIRKIL Exacerbates Cardiac Ischemia/Reperfusion Injury by Interacting With Ku70. Circulation Research, 130(5), e3–e17. https://doi.org/10.1161/circresaha.121.318992
Yaman, O., & Topcu-Tarladacalisir, Y. (2018). L-carnitine counteracts prepubertal exposure to cisplatin induced impaired sperm in adult rats by preventing germ cell apoptosis. Biotechnic & Histochemistry, 1-11. doi:10.1080/10520295.2017.1401661
Yauk, C. L., Lambert, I. B., Meek, M. E. B., Douglas, G. R. & Marchetti, F. (2015). Development of the adverse outcome pathway “alkylation of DNA in male premeiotic germ cells leading to heritable mutations” using the OECD’s users’ handbook supplement. Environmental and Molecular Mutagenesis, 56(9), 724–750. https://doi.org/10.1002/em.21954
Appendix 1
List of MIEs in this AOP
Event: 97: Alkylation, DNA
Short Name: Alkylation, DNA
Event Component
| Process | Object | Action |
|---|---|---|
| DNA alkylation | deoxyribonucleic acid | increased |
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:15 - Alkylation of DNA in male pre-meiotic germ cells leading to heritable mutations | MolecularInitiatingEvent |
| Aop:139 - Alkylation of DNA leading to cancer 1 | MolecularInitiatingEvent |
| Aop:141 - Alkylation of DNA leading to cancer 2 | MolecularInitiatingEvent |
| Aop:322 - Alkylation of DNA leading to decreased sperm count | MolecularInitiatingEvent |
Stressors
| Name |
|---|
| Diethyl nitrosamine |
| Diethyl sulfate |
| Dimethyl nitrosamine |
| Dimethyl sulfate |
| Ethyl methanesulfonate |
| Ethyl nitrosourea |
| Ethyl-N'-nitro-N-nitrosoguanidine |
| Isopropyl methanesulfonate |
| Methyl methanesulfonate |
| Methyl-l-N'-nitro-N-nitroguanidine |
Biological Context
| Level of Biological Organization |
|---|
| Molecular |
Cell term
| Cell term |
|---|
| eukaryotic cell |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| mouse | Mus musculus | High | NCBI |
| Syrian golden hamster | Mesocricetus auratus | High | NCBI |
| rat | Rattus norvegicus | High | NCBI |
| Homo sapiens | Homo sapiens | High | NCBI |
| Sex | Evidence |
|---|---|
| Mixed | High |
Alkylated DNA has been measured in somatic cells in a variety of species, from prokaryotic organisms, to rodents in vivo, to human cells in culture. Theoretically, DNA alkylation can occur in any cell type in any organism.
Key Event Description
The event involves DNA alkylation to form a variety of different DNA adducts (i.e., alkylated nucleotides). Alkylation occurs at various sites in DNA and can include alkylation of adenine- Nl, - N3, - N7, guanine- N3, - O6, - N7, thymine-O2, - N3, - O4, cytosine- O2, -N3, and the phosphate (diester) group (reviewed in detail in Beranek 1990). In addition, alkylation can involve modification with different sizes of alkylation groups (e.g., methyl, ethyl, propyl). It should be noted that many of these adducts are not stable or are readily repaired (discussed in more detail below). A small proportion of adducts are stable and remain bound to DNA for long periods of time.
How it is Measured or Detected
There is no OECD guideline for measurement of alkylated DNA, although technologies for their detection are established. Reviews of modern methods to measure DNA adducts include Himmelstein et al,. 2009 and Philips et al., 2000.
High performance liquid chromatography (HPLC) methods can be used to measure whether an agent is capable of alkylating DNA in somatic cells. Alkyl adducts in somatic cells can be measured using immunological methods (described in Nehls et al. 1984), as well as HPLC (methods in de Groot et al. 1994) or a combination of 32P post-labeling, HPLC and immunologic detection (Kang et al. 1992). We note that mass spectrometry provides structural specificity and confirmation of the structure of DNA adducts.
DNA alkylation can also be measured using a modified comet assay. This method involves the digestion of alkylated DNA bases with 3–methyladenine DNA glycosylase (Collins et al., 2001; Hasplova et al., 2012) followed by the standard comet assay to detect where alkyl adducts occur. The advantage of this method is that the alkaline version of the comet assay, as a core method, has an in vivo OECD guideline.
Finally, structure-activity relationships (SARs) have been developed to predict the possibility that a chemical will alkylate DNA (e.g., Vogel and Ashby, 1994; Benigni, 2005; Dai et al., 1989; Lewis and Griffith, 1987).
Measurement of alkylation in male germ cells:
In rodent testes, studies have detected adducts in situ by immuhistocytological staining. For example, fixed testes are incubated with O6-EtGua -specific mouse monoclonal antibody and subsequently with a labeled anti-mouse IgG F antibody. Nuclear DNA is counterstained with DAPI 4,6-diamidino- 2-phenylindole. Fluorescence signals from immunostained O6-EtGua residues in DNA are visualized by fluorescence microscopy and quantitated using an image analysis system. Methods are described in (Seiler et al. 1997). An immunoslot blot assay for detection of O6-EtGua has been described previously in (Mientjes et al. 1996).
Alternatively, rodents have also been exposed to radio-labeled alkylating agents. Examples from the literature include [2-3H] ENU, [1-3H]di-ethyl sulfate, or [1-3H]ethyl-methane sulfonate. Following treatment with the labeled chemical, testis and other tissues of interest are removed. Germ cells are released from tubuli by pushing out the contents with forceps. Using this procedure all germ-cell stages are liberated from the tubuli, with the possible exception of part of the population of stem-cell spermatogonia that remain attached to the walls of the tubuli. DNA is then extracted from germ cells, empty testis tubuli and other tissues of interest. DNA adduct formation is determined after neutral and acid hydrolysis of DNA followed by separation of the various ethylation products using HPLC (described in van Zeeland et al. 1990).
References
Benigni, R. (2005), "Structure-activity relationship studies of chemical mutagens and carcinogens: mechanistic investigations and prediction and approaches", Chem. Rev., 105: 1767-1800.
Beranek, D.T. (1990), "Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents", Mutation Res., 231: 11-30.
Collins, A.R., M. Dusinská and A. Horská (2001), "A Detection of alkylation damage in human lymphocyte DNA with the comet assay". Acta Biochim Pol., 48: 611-4.
Dai, Q.H. and R.G. Zhong (1989), "Quantitative pattern recognition for structure-carcinogenic activity relationship of N-nitroso compounds based upon Di-region theory", Sci China B., 32:776-790.
de Groot, A.J., J.G. Jansen, C.F. van Valkenburg and A.A. van Zeeland (1994), "Molecular dosimetry of 7-alkyl- and O6-alkylguanine in DNA by electrochemical detection", Mutat Res., 307: 61-6.
Hašplová, K., A. Hudecová, Z. Magdolénová, M. Bjøras, E. Gálová, E. Miadoková and M. Dušinská (2012), "DNA alkylation lesions and their repair in human cells: modification of the comet assay with 3-methyladenine DNA glycosylase (AlkD)", Toxicol Lett., 208: 76-81.
Himmelstein, M.W., P.J. Boogaard, J. Cadet, P.B. Farmer, J.J. Kim, E.A. Martin, R. Persaud and D.E. Shuker (2009), "Creating context for the use of DNA adduct data in cancer risk assessment: II. Overview of methods of identification and quantitation of DNA damage", Crit. Rev. Toxicol., 39: 679-94.
Kamino, K., F. Seiler, M. Emura, J. Thomale, M.F. Rajewsky and U. Mohr (1995), "Formation of O6-ethylguanine in spermatogonial DNA of adult Syrian golden hamster by intraperitoneal injection of diethylnitrosamine", Exp. Toxicol. Pathol., 47: 443-445.
Kang, H.I., C. Konishi, G. Eberle, M.F. Rajewsky, T. Kuroki and N.H. Huh (1992), "Highly sensitive, specific detection of O6-methylguanine, O4-methylthymine, and O4-ethylthymine by the combination of high-performance liquid chromatography prefractionation, 32P postlabeling, and immunoprecipitation", Cancer Res., 52: 5307-5312.
Lewis, D.F. and V.S. Griffiths (1987), "Molecular electrostatic potential energies and methylation of DNA bases: a molecular orbital-generated quantitative structure-activity relationship", Xenobiotica, 17: 769-776.
Mientjes, E.J., K. Hochleitner, A. Luiten-Schuite, J.H. van Delft, J. Thomale, F. Berends, M.F. Rajewsky, P.H. Lohman and R.A. Baan (1996), "Formation and persistence of O6-ethylguanine in genomic and transgene DNA in liver and brain of lambda(lacZ) transgenic mice treated with N-ethyl-N-nitrosourea", Carcinogenesis, 17: 2449-2454.
Nehls, P., M.F. Rajewsky, E. Spiess, D. Werner (1984), "Highly sensitive sites for guanine-O6 ethylation in rat brain DNA exposed to N-ethyl-N-nitrosourea in vivo", EMBO J., 3:327-332.
Phillips, D.H., P.B. Farmer, F.A. Beland, R.G. Nath, M.C. Poirier, M.V. Reddy and K.W. Turteltaub (2000), "Methods of DNA adduct determination and their application to testing compounds for genotoxicity", Environ. Mol. Mutagen., 35: 222-233.
Scherer, E., A.A. Jenner and L. den Engelse (1987), "Immunocytochemical studies on the formation and repair of O6-alkylguanine in rat tissues", IARC Sci. Publ., 84: 55-58.
Sega, G.A., C.R. Rohrer, H.R. Harvey and A.E. Jetton (1986), "Chemical dosimetry of ethyl nitrosourea in the mouse testis", Mutat. Res., 159: 65-74.
Seiler, F., K. Kamino, M. Emura, U. Mohr and J. Thomale (1997), "Formation and persistence of the miscoding DNA alkylation product O6-ethylguanine in male germ cells of the hamster", Mutat. Res., 385: 205-211.
van Zeeland, A.A., A. de Groot and A. Neuhäuser-Klaus (1990), "DNA adduct formation in mouse testis by ethylating agents: a comparison with germ-cell mutagenesis", Mutat. Res. 231: 55-62.
Vogel, E.W., Ashby, J. (1994), "Structure-activity relationships: experimental approaches." In: Methods to asses DNA Damage and repair: Interspecies comparisons. Edited by R.T. Tardiff, P.H.M. Lohman and G.N. Wogan, SCOPE, Wiley and Sons LTD.
List of Key Events in the AOP
Event: 155: Inadequate DNA repair
Short Name: Inadequate DNA repair
Event Component
| Process | Object | Action |
|---|---|---|
| DNA repair | deoxyribonucleic acid | abnormal |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Ionizing Radiation |
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| mouse | Mus musculus | High | NCBI |
| rat | Rattus norvegicus | Moderate | NCBI |
| Syrian golden hamster | Mesocricetus auratus | Moderate | NCBI |
| Homo sapiens | Homo sapiens | High | NCBI |
| cow | Bos taurus | Low | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
The retention of adducts has been directly measured in many different types of eukaryotic somatic cells (in vitro and in vivo). In male germ cells, work has been done on hamsters, rats and mice. The accumulation of mutation and changes in mutation spectrum has been measured in mice and human cells in culture. Theoretically, saturation of DNA repair occurs in every species (prokaryotic and eukaryotic). The principles of this work were established in prokaryotic models. Nagel et al. (2014) have produced an assay that directly measures DNA repair in human cells in culture.
NHEJ is primarily used by vertebrate multicellular eukaryotes, but it also been observed in plants. Furthermore, it has recently been discovered that some bacteria (Matthews et al., 2014) and yeast (Emerson et al., 2016) also use NHEJ. In terms of invertebrates, most lack the core DNA-PKcs and Artemis proteins; they accomplish end joining by using the RA50:MRE11:NBS1 complex (Chen et al., 2001). HR occurs naturally in eukaryotes, bacteria, and some viruses (Bhatti et al., 2016).
Taxonomic applicability: Inadequate DNA repair is applicable to all species, as they all contain DNA (White & Vijg, 2016).
Life stage applicability: This key event is not life stage specific as any life stage can have poor repair, though as individuals age their repair process become less effective (Gorbunova & Seluanov, 2016).
Sex applicability: There is no evidence of sex-specificity for this key event, with initial rate of DNA repair not significantly different between sexes (Trzeciak et al., 2008).
Evidence for perturbation by a stressor: Multiple studies demonstrate that inadequate DNA repair can occur as a result of stressors such as ionizing and non-ionizing radiation, as well as chemical agents (Kuhne et al., 2005; Rydberg et al., 2005; Dahle et al., 2008; Seager et al., 2012; Wilhelm, 2014; O’Brien et al., 2015).
Key Event Description
DNA lesions may result from the formation of DNA adducts (i.e., covalent modification of DNA by chemicals), or by the action of agents such as radiation that may produce strand breaks or modified nucleotides within the DNA molecule. These DNA lesions are repaired through several mechanistically distinct pathways that can be categorized as follows:
- Damage reversal acts to reverse the damage without breaking any bonds within the sugar phosphate backbone of the DNA. The most prominent enzymes associated with damage reversal are photolyases (Sancar, 2003) that can repair UV dimers in some organisms, and O6-alkylguanine-DNA alkyltransferase (AGT) (Pegg 2011) and oxidative demethylases (Sundheim et al., 2008), which can repair some types of alkylated bases.
- Excision repair involves the removal of a damaged nucleotide(s) through cleavage of the sugar phosphate backbone followed by re-synthesis of DNA within the resultant gap. Excision repair of DNA lesions can be mechanistically divided into:
a) Base excision repair (BER) (Dianov and Hübscher, 2013), in which the damaged base is removed by a damage-specific glycosylase prior to incision of the phosphodiester backbone at the resulting abasic site. This leads to an intermediate that contains a DNA strand break, whereby DNA ligase is then recruited to seal the ends of the DNA.
b) Nucleotide excision repair (NER) (Schärer, 2013), in which the DNA strand containing the damaged nucleotide is incised at sites several nucleotides 5’ and 3’ to the site of damage, and a polynucleotide containing the damaged nucleotide is removed prior to DNA resynthesis within the resultant gap and sealing of the ends by DNA ligase.
c) Mismatch repair (MMR) (Li et al., 2016) which does not act on DNA lesions but does recognize mispaired bases resulting from replication errors. In MMR the strand containing the misincorporated base is removed prior to DNA resynthesis.
The major pathway that removes oxidative DNA damage is base excision repair (BER), which can be either monofunctional or bifunctional; in mammals, a specific DNA glycosylase (OGG1: 8-Oxoguanine glycosylase) is responsible for excision of 8-oxoguanine (8-oxoG) and other oxidative lesions (Hu et al., 2005; Scott et al., 2014; Whitaker et al., 2017). We note that long-patch BER is used for the repair of clustered oxidative lesions, which uses several enzymes from DNA replication pathways (Klungland and Lindahl, 1997). These pathways are described in detail in various reviews e.g., (Whitaker et al., 2017).
- Single strand break repair (SSBR) involves different proteins and enzymes depending on the origin of the SSB (e.g., produced as an intermediate in excision repair or due to direct chemical insult) but the same general steps of repair are taken for all SSBs: detection, DNA end processing, synthesis, and ligation (Caldecott, 2014). Poly-ADP-ribose polymerase1 (PARP1) detects and binds unscheduled SSBs (i.e., not deliberately induced during excision repair) and synthesizes PAR as a signal to the downstream factors in repair. PARP1 is not required to initiate SSBR of BER intermediates. The XRCC1 protein complex is then recruited to the site of damage where a common DNA intermediate as BER was generated, and acts as a scaffold for proteins and enzymes required for repair. Depending on the nature of the damaged termini of the DNA strand, different enzymes are required for end processing to generate the substrates that DNA polymerase β (Polβ; short patch repair) or Pol δ/ε (long patch repair) can bind to synthesize over the gap, although end processing is generally done by polynucleotide kinase. Synthesis in long-patch repair displaces a single stranded flap which is excised by flap endonuclease 1 (FEN1). In short-patch repair, the XRCC1/Lig3α complex joins the two ends after synthesis. In long-patch repair, the PCNA/Lig1 complex ligates the ends. (Caldecott, 2014).
- Double strand break repair (DSBR) is necessary to preserve genomic integrity when breaks occur in both strands of a DNA molecule. There are two major pathways for DSBR: homologous recombination (HR), which operates primarily during the S phase of dividing cell types, and nonhomologous end joining (NHEJ), which can function in both dividing and non-dividing cell types. No repair occurs in the M phase (Teruaki Iyama and David M. Wilson III, 2013). DNA repair in mitosis is controversial (Mladenov et al., 2023).
Complex lesions can be created by a single mutagen and can be more difficult to repair, as the mechanism behind how different repair pathways cooperate to address this is still unclear (Aleksandrov et al., 2018). In higher eukaryotes such as mammals, NHEJ is usually the preferred pathway for DNA DSBR. Its use, however, is dependent on the cell type, the gene locus, and the nuclease platform (Miyaoka et al., 2016). The use of NHEJ is also dependent on the cell cycle; NHEJ is generally not the pathway of choice when the cell is in the late S or G2 phase of the cell cycle, or in mitotic cells when the sister chromatid is directly adjacent to the double-strand break (DSB) (Lieber et al., 2003). In these cases, the HR pathway is commonly used for repair of DSBs. Despite this, NHEJ is still used more commonly than HR in human cells. Classical NHEJ (C-NHEJ) is the most common NHEJ repair mechanism, but alternative NHEJ (alt-NHEJ) can also occur, especially in the absence of C-NHEJ and HR.
The process of C-NHEJ in humans requires at least seven core proteins: Ku70, Ku86, DNA-dependent protein kinase complex (DNA-PKcs ), Artemis, X-ray cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF), and DNA ligase IV (Boboila et al., 2012). When DSBs occur, the Ku proteins, which have a high affinity for DNA ends, will bind to the break site and form a heterodimer. This protects the DNA from exonucleolytic attack and acts to recruit DNA-PKcs, the catalytic subunit, thus forming a trimeric complex on the ends of the DNA strands. Alternative NHEJ, or alt NHEJ, uses small similar sequences in two broken DNA ends to join them together. Unlike the usual repair method (cNHEJ), aNHEJ doesn't need specific proteins like LIG4 and KU. Instead, it relies on the MRN complex to process the breaks. However, alt NHEJ tends to cause mutations by adding or removing bits of DNA during the repair (Chaudhuri and Nussenzweig, 2017). The kinase activity of DNA-PKcs is then triggered, causing DNA-PKcs to auto-phosphorylate and thereby lose its kinase activity; the now phosphorylated DNA-PKcs dissociates from the DNA-bound Ku proteins. The free DNA-PKcs phosphorylates Artemis, an enzyme that possesses 5’-3’ exonuclease and endonuclease activity in the presence of DNA-PKcs and ATP. Artemis is responsible for ‘cleaning up’ the ends of the DNA. For 5’ overhangs, Artemis nicks the overhang, generally leaving a blunt duplex end. For 3’ overhangs, Artemis will often leave a four- or five-nucleotide single stranded overhang (Pardo et al., 2009; Fattah et al., 2010; Lieber et al., 2010). Next, the XLF and XRCC4 proteins form a complex which makes a channel to bind DNA and aligns the ends for efficient ligation via DNA ligase IV (Hammel et al., 2011).
The process of alt-NHEJ is less well understood than C-NHEJ and is a lower fidelity mechanism. Alt-NHEJ is known to involve slightly different core proteins than C-NHEJ and required microhomology repeats, but the steps of the pathway are essentially the same between the two processes (reviewed in Chiruvella et al., 2013). It is established, however, that alt-NHEJ is more error-prone in nature than C-NHEJ, which contributes to incorrect DNA repair. Alt-NHEJ is thus considered primarily to be a backup repair mechanism (reviewed in Chiruvella et al., 2013).
In contrast to NHEJ, HR takes advantage of similar or identical DNA sequences to repair DSBs and is not error-prone (Sung and Klein, 2006). The initiating step of HR is the creation of a 3’ single strand DNA (ss-DNA) overhang. Combinases such as RecA and Rad51 then bind to the ss-DNA overhang, and other accessory factors, including Rad54, help recognize and invade the homologous region on another DNA strand. From there, DNA polymerases are able to elongate the 3’ invading single strand and resynthesize the broken DNA strand using the corresponding sequence on the homologous strand.
Fidelity of DNA Repair
Most DNA repair pathways are extremely efficient. However, in principal, all DNA repair pathways can be overwhelmed when the DNA lesion burden exceeds the capacity of a given DNA repair pathway to recognize and remove the lesion. Exceeded repair capacity may lead to toxicity or mutagenesis following DNA damage. Apart from extremely high DNA lesion burden, inadequate repair may arise through several different specific mechanisms. For example, during repair of DNA containing O6-alkylguanine adducts, AGT irreversibly binds a single O6-alkylguanine lesion and as a result is inactivated (this is termed suicide inactivation, as its own action causes it to become inactivated). Thus, the capacity of AGT to carry out alkylation repair can become rapidly saturated when the DNA repair rate exceeds the de novo synthesis of AGT (Pegg, 2011).
A second mechanism relates to cell specific differences in the cellular levels or activity of some DNA repair proteins. For example, XPA is an essential component of the NER complex. The level of XPA that is active in NER is low in the testes, which may reduce the efficiency of NER in testes as compared to other tissues (Köberle et al., 1999). Likewise, both NER and BER have been reported to be deficient in cells lacking functional p53 (Adimoolam and Ford, 2003; Hanawalt et al., 2003; Seo and Jung, 2004). A third mechanism relates to the importance of the DNA sequence context of a lesion in its recognition by DNA repair enzymes. For example, 8-oxoguanine (8-oxoG) is repaired primarily by BER; the lesion is initially acted upon by a bifunctional glycosylase, OGG1, which carries out the initial damage recognition and excision steps of 8-oxoG repair. However, the rate of excision of 8-oxoG is modulated strongly by both chromatin components (Menoni et al., 2012) and DNA sequence context (Allgayer et al., 2013) leading to significant differences in the repair of lesions situated in different chromosomal locations.
DNA repair is also remarkably error-free. However, misrepair can arise during repair under some circumstances. DSBR is notably error prone, particularly when breaks are processed through NHEJ, during which partial loss of genome information is common at the site of the double strand break (Iyama and Wilson, 2013). This is because NHEJ rejoins broken DNA ends without the use of extensive homology; instead, it uses the microhomology present between the two ends of the DNA strand break to ligate the strand back into one. When the overhangs are not compatible, however, indels (insertion or deletion events), duplications, translocations, and inversions in the DNA can occur. These changes in the DNA may lead to significant issues within the cell, including alterations in the gene determinants for cellular fatality (Moore et al., 1996).
Activation of mutagenic DNA repair pathways to withstand cellular or replication stress either from endogenous or exogenous sources can promote cellular viability, albeit at a cost of increased genome instability and mutagenesis (Fitzgerald et al., 2017). These salvage DNA repair pathways including, Break-induced Replication (BIR) and Microhomology-mediated Break-induced Replication (MMBIR). BIR repairs one-ended DSBs and has been extensively studied in yeast as well as in mammalian systems. BIR and MMBIR are linked with heightened levels of mutagenesis, chromosomal rearrangements and ensuing genome instability (Deem et al., 2011; Sakofsky et al., 2015; Saini et al., 2017; Kramara et al., 2018). In mammalian genomes BIR-like synthesis has been proposed to be involved in late stage Mitotic DNA Synthesis (MiDAS) that predominantly occurs at so-called Common Fragile Sites (CFSs) and maintains telomere length under s conditions of replication stress that serve to promote cell viability (Minocherhomji et al., 2015; Bhowmick et al., 2016; Dilley et al., 2016).
Misrepair may also occur through other repair pathways. Excision repair pathways require the resynthesis of DNA and rare DNA polymerase errors during gap resynthesis will result in mutations (Brown et al., 2011). Errors may also arise during gap resynthesis when the strand that is being used as a template for DNA synthesis contains DNA lesions (Kozmin and Jinks-Robertson, 2013). In addition, it has been shown that sequences that contain tandemly repeated sequences, such as CAG triplet repeats, are subject to expansion during gap resynthesis that occurs during BER of 8-oxoG damage (Liu et al., 2009).
How it is Measured or Detected
There is no test guideline for this event. The event is usually inferred from measuring the retention of DNA adducts or the creation of mutations as a measure of lack of repair or incorrect repair. These ‘indirect’ measures of its occurrence are crucial to determining the mechanisms of genotoxic chemicals and for regulatory applications (i.e., determining the best approach for deriving a point of departure). More recently, a fluorescence-based multiplex flow-cytometric host cell reactivation assay (FM-HCR) has been developed to directly measure the ability of human cells to repair plasmid reporters (Nagel et al., 2014).
Indirect Measurement
In somatic and spermatogenic cells, measurement of DNA repair is usually inferred by measuring DNA adduct formation/removal. Insufficient repair is inferred from the retention of adducts and from increasing adduct formation with dose. Insufficient DNA repair is also measured by the formation of increased numbers of mutations and alterations in mutation spectrum. The methods will be specific to the type of DNA adduct that is under study.
Some EXAMPLES are given below for alkylated DNA.
DOSE-RESPONSE CURVE FOR ALKYL ADDUCTS/MUTATIONS: It is important to consider that some adducts are not mutagenic at all because they are very effectively repaired. Others are effectively repaired, but if these repair processes become overwhelmed mutations begin to occur. The relationship (shape of dose-response curve) between exposure to mutagenic agents and mutations provide an indication of whether the removal of adducts occurs, and whether it is more efficient at low doses. Sub-linear dose-response curves (hockey stick or j-shape curves) for mutation induction indicates that adducts are not converted to mutations at low doses. This suggests the effective repair of adducts at low doses, followed by saturation of repair at higher doses (Clewell et al., 2019). Thus, measurement of a clear point of inflection in the dose-response curve for mutations suggests that repair does occur, at least to some extent, at low dosees but that reduced repair efficiency arises above the inflection point. A lack of increase in mutation frequencies (i.e., flat line for dose-response) for a compound showing a dose-dependent increase in adducts would imply that the adducts formed are either not mutagenic or are effectively repaired.
RETENTION OF ALKYL ADDUCTS: Alkylated DNA can be found in cells long after exposure has occurred. This indicates that repair has not effectively removed the adducts. For example, DNA adducts have been measured in hamster and rat spermatogonia several days following exposure to alkylating agents, indicating lack of repair (Seiler et al., 1997; Scherer et al., 1987).
MUTATION SPECTRUM: Shifts in mutation spectrum (i.e., the specific changes in the DNA sequence) following a chemical exposure (relative to non-exposed mutation spectrum) indicates that repair was not operating effectively to remove specific types of lesions. The shift in mutation spectrum is indicative of the types of DNA lesions (target nucleotides and DNA sequence context) that were not repaired. For example, if a greater proportion of mutations occur at guanine nucleotides in exposed cells, it can be assumed that the chemical causes DNA adducts on guanine that are not effectively repaired.
Direct Measurement
Nagel et al. (2014) we developed a fluorescence-based multiplex flow-cytometric host cell reactivation assay (FM-HCR) to measures the ability of human cells to repair plasmid reporters. These reporters contain different types and amounts of DNA damage and can be used to measure repair through by NER, MMR, BER, NHEJ, HR and MGMT.
Please refer to the table below for additional details and methodologies for detecting DNA damage and repair.
| Assay Name | References | Description | DNA Damage/Repair Being Measured | OECD Approved Assay |
| Dose-Response Curve for Alkyl Adducts/ Mutations |
Lutz 1991
Clewell 2016 |
Creation of a curve plotting the stressor dose and the abundance of adducts/mutations; Characteristics of the resulting curve can provide information on the efficiency of DNA repair |
Alkylation, oxidative damage, or DSBs |
N/A |
| Retention of Alkyl Adducts |
Seiler 1997
Scherer 1987 |
Examination of DNA for alkylation after exposure to an alkylating agent; Presence of alkylation suggests a lack of repair | Alkylation | N/A |
| Mutation Spectrum | Wyrick 2015 | Shifts in the mutation spectrum after exposure to a chemical/mutagen relative to an unexposed subject can provide an indication of DNA repair efficiency, and can inform as to the type of DNA lesions present |
Alkylation, oxidative damage, or DSBs |
N/A |
| DSB Repair Assay (Reporter constructs) | Mao et al., 2011 | Transfection of a GFP reporter construct (and DsRed control) where the GFP signal is only detected if the DSB is repaired; GFP signal is quantified using fluorescence microscopy or flow cytometry | DSBs | N/A |
| Primary Rat Hepatocyte DNA Repair Assay |
Jeffrey and Williams, 2000
Butterworth et al., 1987 |
Rat primary hepatocytes are cultured with a 3H-thymidine solution in order to measure DNA synthesis in response to a stressor in non-replicating cells; Autoradiography is used to measure the amount of 3H incorporated in the DNA post-repair | Unscheduled DNA synthesis in response to DNA damage | N/A |
| Repair synthesis measurement by 3H-thymine incorporation | Iyama and Wilson, 2013 | Measure DNA synthesis in non-dividing cells as indication of gap filling during excision repair | Excision repair | N/A |
| Comet Assay with Time-Course |
Olive et al., 1990
Trucco et al., 1998 - Dunkenberger et al., 2022 |
Comet assay is performed with a time-course under alkaline conditions to detect SSBs and DSBs. Quantity of DNA in the tail should decrease as DNA repair progresses | DSBs | Yes (No. 489) |
| Flow Cytometry | Corneo et al., 2007 | The alt-NHEJ flow cytometer method involves utilizing an extrachromosomal substrate. Green fluorescent protein (GFP) expression is indicative of successful alt-NHEJ activity, contingent on the removal of 10 nucleotides from each end of the DNA and subsequent rejoining within a 9-nucleotide microhomology region. This approach provides a quantitative and visual means to measure the efficiency of alternative non-homologous end joining in cellular processes. | Alt NHEJ | No |
| Pulsed Field Gel Electro-phoresis (PFGE) with Time-Course | Biedermann et al., 1991 | PFGE assay with a time-course; Quantity of small DNA fragments should decrease as DNA repair progresses | DSBs | N/A |
|
Fluorescence -Based Multiplex Flow-Cytometric Host Reactivation Assay (FM-HCR) |
Nagel et al., 2014 | Measures the ability of human cells to repair plasma reporters, which contain different types and amounts of DNA damage; Used to measure repair processes including HR, NHEJ, BER, NER, MMR, and MGMT | HR, NHEJ, BER, NER, MMR, or MGMT | N/A |
| Alkaline Unwinding Assay with Time Course | Nacci et al. 1991 | DNA is stored in alkaline solutions with DNA-specific dye and allowed to unwind following removal from tissue, increased strand damage associated with increased unwinding. Samples analyzed at different time points to compare remaining damage following repair opportunities | DSBs | Yes (No. 489) |
| Sucrose Density Gradient Centrifugation with Time Course | Larsen et al. 1982 | Strand breaks alter the molecular weight of the DNA piece. DNA in alkaline solution centrifuged into sugar density gradient, repeated set time apart. The less DNA breaks identified in the assay repeats, the more repair occurred | SSBs | N/A |
| y-H2AX Foci Staining with Time Course |
Mariotti et al. 2013 Penninckx et al. 2021 |
Histone H2AX is phosphorylated in the presence of DNA strand breaks, the rate of its disappearance over time is used as a measure of DNA repair | DSBs | N/A |
| Alkaline Elution Assay with Time Course | Larsen et al. 1982 | DNA with strand breaks elute faster than DNA without, plotted against time intervals to determine the rate at which strand breaks repair | SSBs | N/A |
| 53BP1 foci Detection with Time Course | Penninckx et al. 2021 | 53BP1 is recruited to the site of DNA damage, the rate at which its level decreases over time is used to measure DNA repair | DSBs | N/A |
References
Adimoolam, S. & J.M. Ford (2003), "p53 and regulation of DNA damage recognition during nucleotide excision repair" DNA Repair (Amst), 2(9): 947-54.
Aleksandrov, Radoslav et al. (2018), “Protein Dynamics in Complex DNA Lesions.” Molecular cell,69(6): 1046-1061.e5. doi:10.1016/j.molcel.2018.02.016
Allgayer, J. et al. (2013), "Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence", Nucleic Acids Res, 41(18): 8559-8571. Doi: 10.1093/nar/gkt620.
Beranek, D.T. (1990), "Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents", Mutation Research, 231(1): 11-30. Doi: 10.1016/0027-5107(90)90173-2.
Bhatti, A. et al., (2016), “Homologous Recombination Biology.”, Encyclopedia Britannica.
Bhowmick, R., S. et al. (2016), "RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress", Mol Cell, 64:1117-1126. Doi: 10.1016/j.molcel.2016.10.037.
Biedermann, A. K. et al. (1991), “SCID mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair”, Cell Biology, 88(4): 1394-7. Doi: 10.1073/pnas.88.4.1394.
Boboila, C., F. W. Alt & B. Schwer. (2012), “Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks.” Adv Immunol, 116, 1-49. doi:10.1016/B978-0-12-394300-2.00001-6
Bronstein, S.M. et al. (1991), "Toxicity, mutagenicity, and mutational spectra of N-ethyl-N-nitrosourea in human cell lines with different DNA repair phenotypes", Cancer Research, 51(19): 5188-5197.
Bronstein, S.M. et al. (1992), "Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells", Cancer Research, 52(7): 2008-2011.
Brown, J.A. et al. (2011), "Efficiency and fidelity of human DNA polymerases λ and β during gap-filling DNA synthesis", DNA Repair (Amst)., 10(1):24-33.
Butterworth, E. B. et al., (1987), A protocol and guide for the in vitro rat hepatocyte DNA-repair assay. Mutation Research. 189, 113-21. Doi: 10.1016/0165-1218(87)90017-6.
Caldecott, K. W. (2014), "DNA single-strand break repair", Exp Cell Res, 329(1): 2-8.
Chaudhuri, R.A. and Nussenzweig, A. (2017), “The multifaceted roles of PARP1 in DNA repair and chromatin remodelling”. Nat Rev Mol Cell Biol 18, 610–621. https://doi.org/10.1038/nrm.2017.53
Chen, L. et al., (2001), Promotion of DNA ligase IV-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 8(5), 1105-15.
Chiruvella, K. K., Z. Liang & T. E. Wilson, (2013), Repair of Double-Strand Breaks by End Joining. Cold Spring Harbor Perspectives in Biology, 5(5):127-57. Doi: 10.1101/cshperspect.a012757.
Clewell, R. A. et al. (2019). “Dose-dependence of chemical carcinogenicity: Biological mechanisms for thresholds and implications for risk assessment”. Chem Biol Interact. 2019 Mar 1;301:112-127. doi: 10.1016/j.cbi.2019.01.025.
Corneo, B. et al., 2007, "Rag mutations reveal robust alternative end joining”. Nature 449, 483–486 (2007). https://doi.org/10.1038/nature06168
Dahle, J., et al. (2008), “Overexpression of human OGG1 in mammalian cells decreases ultraviolet A induced mutagenesis”, Cancer Letters, Vol.267, Elsevier, Amsterdam, https://doi.org/10.1016/j.canlet.2008.03.002.
Deem, A. et al. (2011), "Break-Induced Replication Is Highly Inaccurate.", PLoS Biol. 9:e1000594. Doi: 10.1371/journal.pbio.1000594.
Dianov, G.L. & U. Hübscher (2013), "Mammalian base excision repair: the forgotten archangel", Nucleic Acids Res., 41(6):3483-90. Doi: 10.1093/nar/gkt076.
Dilley, R.L. et al. Greenberg (2016), "Break-induced telomere synthesis underlies alternative telomere maintenance", Nature, 539:54-58. Doi: 10.1038/nature20099.
Douglas, G.R. et al. (1995), "Temporal and molecular characteristics of mutations induced by ethylnitrosourea in germ cells isolated from seminiferous tubules and in spermatozoa of lacZ transgenic mice", Proceedings of the National Academy of Sciences of the United States of America, 92(16):7485-7489. Doi: 10.1073/pnas.92.16.7485.
Dunkenberger, Logan et al. (2022), “Comet Assay for the Detection of Single and Double-Strand DNA Breaks.” Methods in molecular biology (Clifton, N.J.), 2422: 263-269. doi:10.1007/978-1-0716-1948-3_18
Fattah, F. et al., (2010), Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet, 6(2), doi:10.1371/journal.pgen.1000855
Fitzgerald, D.M., P.J. Hastings, and S.M. Rosenberg (2017), "Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance", Ann Rev Cancer Biol, 1:119-140. Doi: 10.1146/annurev-cancerbio-050216-121919.
Gorbunova, V. and A. Seluanov. (2016), “DNA double strand break repair, aging and the chromatin connection”, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Vol.788/1-2, Elsevier, Amsterdam, http://dx.doi.org/10.1016/j.mrfmmm.2016.02.004.
Hammel, M. et al., (2011), XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem, 286(37), 32638-32650. doi:10.1074/jbc.M111.272641.
Hanawalt, P.C., J.M. Ford and D.R. Lloyd (2003), "Functional characterization of global genomic DNA repair and its implications for cancer", Mutation Research, 544(2-3): 107–114.
Harbach, P. R. et al., (1989), “The in vitro unscheduled DNA synthesis (UDS) assay in rat primary hepatocytes”, Mutation Research, 216(2):101-10. Doi:10.1016/0165-1161(89)90010-1.
Iyama, T. and D.M. Wilson III (2013), "DNA repair mechanisms in dividing and non-dividing cells", DNA Repair, 12(8): 620– 636.
Jeffrey, M. A.& M. G. Williams, (2000), “Lack of DNA-damaging Activity of Five Non-nutritive Sweeteners in the Rat Hepatocyte/DNA Repair Assay”, Food and Chemical Toxicology, 38: 335-338. Doi: 10.1016/S0278-6915(99)00163-5.
Köberle, B. et al. (1999), "Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours", Curr. Biol., 9(5):273-6. Doi: 10.1016/s0960-9822(99)80118-3.
Kozmin, S.G. & S. Jinks-Robertson S. (2013), “The mechanism of nucleotide excision repair-mediated UV-induced mutagenesis in nonproliferating cells”, Genetics, 193(3): 803-17. Doi: 10.1534/genetics.112.147421.
Kramara, J., B. Osia, and A. Malkova (2018), "Break-Induced Replication: The Where, The Why, and The How", Trends Genet, 34:518-531. Doi: 10.1016/j.tig.2018.04.002.
Kuhne, M., G. Urban and M. Lo (2005), "DNA Double-Strand Break Misrejoining after Exposure of Primary Human Fibroblasts to CK Characteristic X Rays, 29 kVp X Rays and 60Co γ Rays", Radiation. Research, Vol.164/5, Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR3461.1.
Larsen, K.H. et al. (1982), “DNA repair assays as tests for environmental mutagens: A report of the U.S. EPA gene-tox program”, Mutation Research, Vol.98/3, Elsevier, Amsterdam, https://doi.org/10.1016/0165-1110(82)90037-9.
Li Z, A. H. Pearlman, and P. Hsieh (2016), "DNA mismatch repair and the DNA damage response", DNA Repair (Amst), 38:94-101.
Lieber, M. R., (2010), “The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway.” Annu Rev Biochem. 79:181-211. doi:10.1146/annurev.biochem.052308.093131.
Lieber, M. R. et al., (2003), “Mechanism and regulation of human non-homologous DNA end-joining”, Nat Rev Mol Cell Biol. 4(9):712-720. doi:10.1038/nrm1202.
Liu, Y. et al. (2009), "Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion", J. Biol. Chem., 284(41): 28352-28366. Doi: 10.1074/jbc.M109.050286.
Mao, Z. et al., (2011), “SIRT6 promotes DNA repair under stress by activating PARP1”, Science. 332(6036): 1443-1446. doi:10.1126/science.1202723.
Mariotti, L.G. et al. (2013), “Use of the γ-H2AX Assay to Investigate DNA Repair Dynamics Following Multiple Radiation Exposures”, PLoS ONE, Vol.8/11, PLoS, San Francisco, https://doi.org/10.1371/journal.pone.0079541.
Matthews, L. A., & L. A. Simmons, (2014), “Bacterial nonhomologous end joining requires teamwork”, J Bacteriol. 196(19): 3363-3365. doi:10.1128/JB.02042-14.
Menoni, H. et al. (2012), "Base excision repair of 8-oxoG in dinucleosomes", Nucleic Acids Res. ,40(2): 692-700. Doi: 10.1093/nar/gkr761.
Minocherhomji, S. et al. (2015), "Replication stress activates DNA repair synthesis in mitosis", Nature, 528:286-290. Doi: 10.1038/nature16139.
Miyaoka, Y. et al., (2016), “Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing”, Sci Rep, 6, 23549. doi:10.1038/srep23549/.
Mladenov. et al. (2023), . “New Facets of DNA Double Strand Break Repair: Radiation Dose as Key Determinant of HR versus c-NHEJ Engagement”. International journal of molecular sciences, 24(19), 14956. https://doi.org/10.3390/ijms241914956
Moore, J. K., & J. E. Haber, (1996), “Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae”, Molecular and Cellular Biology, 16(5), 2164–73. Doi: 10.1128/MCB.16.5.2164.
Nacci, D. et al. (1992), “Application of the DNA alkaline unwinding assay to detect DNA strand breaks in marine bivalves”, Marine Environmental Research, Vol.33/2, Elsevier BV, Amsterdam, https://doi.org/10.1016/0141-1136(92)90134-8.
Nagel, Z.D. et al. (2014), "Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis", Proc. Natl. Acad. Sci. USA, 111(18):E1823-32. Doi: 10.1073/pnas.1401182111.
O’Brien, J.M. et al. (2015), "Sublinear response in lacZ mutant frequency of Muta™ Mouse spermatogonial stem cells after low dose subchronic exposure to N-ethyl-N-nitrosourea", Environ. Mol. Mutagen., 56(4): 347-55. Doi: 10.1002/em.21932.
Olive, L. P., J. P. Bnath & E. R. Durand, (1990), “Heterogeneity in Radiation-Induced DNA Damage and Repairing Tumor and Normal Cells Measured Using the "Comet" Assay”, Radiation Research. 122: 86-94. Doi: 10.1667/rrav04.1.
Pardo, B., B. Gomez-Gonzalez & A. Aguilera, (2009), “DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship”, Cell Mol Life Sci, 66(6), 1039-1056. doi:10.1007/s00018-009-8740-3.
Pegg, A.E. (2011), "Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools", Chem. Res. Toxicol., 4(5): 618-39. Doi: 10.1021/tx200031q.
Penninckx, S. et al. (2021), “Quantification of radiation-induced DNA double strand break repair foci to evaluate and predict biological responses to ionizing radiation”, NAR Cancer, Vol.3/4, Oxford University Press, Oxford, https://doi.org/10.1093/narcan/zcab046.
Rydberg, B. et al. (2005), "Dose-Dependent Misrejoining of Radiation-Induced DNA Double-Strand Breaks in Human Fibroblasts: Experimental and Theoretical Study for High- and Low-LET Radiation", Radiation Research, Vol.163/5, Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR3346.
Sancar, A. (2003), "Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors", Chem Rev., 103(6): 2203-37. Doi: 10.1021/cr0204348.
Saini, N. et al. (2017), "Migrating bubble during break-induced replication drives conservative DNA synthesis", Nature, 502:389-392. Doi: 10.1038/nature12584.
Sakofsky, C.J. et al. (2015), "Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements", Mol Cell, 60:860-872. Doi: 10.1016/j.molcel.2015.10.041.
Schärer, O.D. (2013), "Nucleotide excision repair in eukaryotes", Cold Spring Harb. Perspect. Biol., 5(10): a012609. Doi: 10.1101/cshperspect.a012609.
Scherer, E., A.A. Jenner and L. den Engelse (1987), "Immunocytochemical studies on the formation and repair of O6-alkylguanine in rat tissues", IARC Sci Publ., 84: 55-8.
Seiler, F., K. Kamino, M. Emura, U. Mohr and J. Thomale (1997), "Formation and persistence of the miscoding DNA alkylation product O6-ethylguanine in male germ cells of the hamster", Mutat Res., 385(3): 205-211. Doi: 10.1016/s0921-8777(97)00043-8.
Shelby, M.D. and K.R. Tindall (1997), "Mammalian germ cell mutagenicity of ENU, IPMS and MMS, chemicals selected for a transgenic mouse collaborative study", Mutation Research, 388(2-3): 99-109. Doi: 10.1016/s1383-5718(96)00106-4.
Seo, Y.R. and H.J. Jung (2004), "The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER)", Exp. Mol. Med., 36(6): 505-509. Doi: 10.1038/emm.2004.64.
Sundheim, O. et al. (2008), "AlkB demethylases flip out in different ways", DNA Repair (Amst)., 7(11): 1916-1923. Doi: 10.1016/j.dnarep.2008.07.015.
Sung, P., & H. Klein, (2006), “Mechanism of homologous recombination: mediators and helicases take on regulatory functions”, Nat Rev Mol Cell Biol, 7(10), 739-750. Doi:10. 1038/nrm2008.
Trucco, C., et al., (1998), “DNA repair defect i poly(ADP-ribose) polymerase-deficient cell lines”, Nucleic Acids Research. 26(11): 2644–2649. Doi: 10.1093/nar/26.11.2644.
Trzeciak, A.R. et al. (2008), “Age, sex, and race influence single-strand break repair capacity in a human population”, Free Radical Biology & Medicine, Vol. 45, Elsevier, Amsterdam, https://doi.org/10.1016/j.freeradbiomed.2008.08.031.
White, R.R. and J. Vijg. (2016), “Do DNA Double-Strand Breaks Drive Aging?”, Molecular Cell, Vol.63, Elsevier, Amsterdam, http://doi.org/10.1016/j.molcel.2016.08.004.
Wyrick, J.J. & S. A. Roberts, (2015), “Genomic approaches to DNA repair and mutagenesis”, DNA Repair (Amst). 36:146-155. doi: 10.1016/j.dnarep.2015.09.018.
van Zeeland, A.A., A. de Groot and A. Neuhäuser-Klaus (1990), "DNA adduct formation in mouse testis by ethylating agents: a comparison with germ-cell mutagenesis", Mutat. Res., 231(1): 55-62.
Event: 1635: Increase, DNA strand breaks
Short Name: Increase, DNA strand breaks
Event Component
| Process | Object | Action |
|---|---|---|
| DNA Strand Break | Deoxyribonucleic acid | increased |
AOPs Including This Key Event
Stressors
| Name |
|---|
| Ionizing Radiation |
| Topoisomerase inhibitors |
| Radiomimetic compounds |
Biological Context
| Level of Biological Organization |
|---|
| Molecular |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| human and other cells in culture | human and other cells in culture | NCBI |
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
Taxonomic applicability: DNA strand breaks are relevant to all species, including vertebrates such as humans, that contain DNA (Cannan & Pederson, 2016).
Life stage applicability: This key event is not life stage specific as all life stages display strand breaks. However, there is an increase in baseline levels of DNA strand breaks seen in older individuals though it is unknown whether this change due to increased break induction or a greater retention of breaks due to poor repair (White & Vijg, 2016).
Sex applicability: This key event is not sex specific as both sexes display evidence of strand breaks. In some cell types, such as peripheral blood mononuclear cells, males show higher levels of single strand breaks than females (Garm et al., 2012).
Evidence for perturbation by a stressor: There are studies demonstrating that increased DNA strand breaks can result from exposure to multiple stressor types including ionizing & non-ionizing radiation, chemical agents, and oxidizing agents (EPRI, 2014; Hamada, 2014; Cencer et al., 2018; Cannan & Pederson, 2016; Yang et al., 1998).
Key Event Description
DNA strand breaks are a type of damage resulting from the hydrolysis of phosphodiester groups in the backbone of DNA molecules (Gates, 2009) and can occur on a single strand (single strand breaks; SSBs) or both strands (double strand breaks; DSBs). SSBs arise when the sugar phosphate backbones connecting adjacent nucleotides in DNA are simultaneously hydrolyzed such that the hydrogen bonds between complementary bases are not able to hold the two strands together. DSBs are generated when both strands are simultaneously broken at sites that are sufficiently close to one another that base-pairing and chromatin structure are insufficient to keep the two DNA ends juxtaposed. As a consequence, the two DNA ends generated by a DSB can physically dissociate from one another, becoming difficult to repair and increasing the chance of inappropriate recombination with other sites in the genome (Jackson, 2002). SSB can turn into DSB if the replication fork stalls at the lesion leading to fork collapse. Strand breaks are intermediates in various biological events, including DNA repair (e.g., excision repair), as well as other normal cellular processes where DSBs act as genetic shufflers to generate genetic diversity for V(D)J recombination in lymphoid cells, and chromatin remodeling in both somatic cells and germ cells, and meiotic recombination in gametes.
Strand breaks are intermediates in various biological events, including DNA repair (e.g., excision repair), V(D)J recombination in developing lymphoid cells and chromatin remodeling in both somatic cells and germ cells. The spectrum of damage can be complex, particularily if the stressor is from large amounts of deposited energy which can result in complex lesions and clustered damage defined as two or more oxidized bases, abasic sites or strand breaks on opposing DNA strands within a few helical turns. These lesions are more difficult to repair and have been studied in many types of models (Barbieri et al., 2019 and Asaithamby et al., 2011). DSBs and complex lesions are of particular concern, as they are considered the most lethal and deleterious type of DNA lesion. If misrepaired or left unrepaired, DSBs may drive the cell towards genomic instability, apoptosis or tumorigenesis (Beir, 1999).
How it is Measured or Detected
Please refer to the table below for details regarding these and other methodologies for detecting DNA DSBs.
|
Method of Measurement |
References |
Description |
OECD Approved Method? |
|
Comet Assay (Single Cell Gel Eletrophoresis - Alkaline) |
Collins, 2004; Olive and Banath, 2006; Platel et al., 2011; Nikolova et al., 2017 |
To detect SSBs or DSBs, single cells are encapsulated in agarose on a slide, lysed, and subjected to gel electrophoresis at an alkaline pH (pH >13); DNA fragments are forced to move, forming a "comet"-like appearance |
Yes |
|
γ-H2AX Foci Quantification - Flow Cytometry |
Rothkamm and Horn, 2009; Bryce et al., 2016 |
Measurement of γ-H2AX immunostaining in cells by flow cytometry, normalized to total levels of H2AX |
No |
|
γ-H2AX Foci Quantification - Western Blot |
Burma et al., 2001; Revet et al., 2011 |
Measurement of γ-H2AX immunostaining in cells by Western blotting, normalized to total levels of H2AX |
No |
|
γ-H2AX Foci Quantification - Microscopy |
Redon et al., 2010; Mah et al., 2010; Garcia-Canton et al., 2013 |
Quantification of γ-H2AX immunostaining by counting γ-H2AX foci visualized with a microscope |
No |
|
γ-H2AX Foci Quantification - ELISA |
Ji et al., 2017 |
Measurement of γ-H2AX in cells by ELISA, normalized to total levels of H2AX |
No |
|
Pulsed Field Gel Electrophoresis (PFGE) |
Ager et al., 1990; Gardiner et al., 1985; Herschleb et al., 2007; Kawashima et al., 2017 |
To detect DSBs, cells are embedded and lysed in agarose, and the released DNA undergoes gel electrophoresis in which the direction of the voltage is periodically alternated; Large DNA fragments are thus able to be separated by size |
No |
|
The TUNEL (Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling) Assay |
Loo, 2011 |
To detect strand breaks, dUTPs added to the 3’OH end of a strand break by the DNA polymerase terminal deoxynucleotidyl transferase (TdT) are tagged with a fluorescent dye or a reporter enzyme to allow visualization |
No |
|
In Vitro DNA Cleavage Assays using Topoisomerase |
Nitiss, 2012 |
Cleavage of DNA can be achieved using purified topoisomerase; DNA strand breaks can then be separated and quantified using gel electrophoresis |
No |
|
PCR assay |
Figueroa‑González & Pérez‑Plasencia, 2017 |
Assay of strand breaks through the observation of DNA amplification prevention. Breaks block Taq polymerase, reducing the number of DNA templates, preventing amplification |
No |
|
Sucrose density gradient centrifuge |
Raschke et al. 2009 |
Division of DNA pieces by density, increased fractionation leads to lower density pieces, with the use of a sucrose cushion |
No |
|
Alkaline Elution Assay |
Kohn, 1991 |
Cells lysed with detergent-solution, filtered through membrane to remove all but intact DNA |
No |
|
Unwinding Assay |
Nacci et al. 1992 |
DNA is stored in alkaline solutions with DNA-specific dye and allowed to unwind following removal from tissue, increased strand damage associated with increased unwinding |
Yes |
|
STRIDE assay |
Zilio and Ulrich, 2021 |
STRIDE (SensiTive Recognition of Individual DNA Ends) combines in situ nick translation with the proximity ligation assay (PLA) to detect single-strand breaks (sSTRIDE) or double-strand breaks (dSTRIDE). In this process, lesions labeled through nick translation with biotinylated nucleotides are identified by a PLA signal, which arises from the interaction of two anti-biotin antibodies from different species.
|
No |
|
sBLISS |
Bouwmann et al. 2020 |
sBLISS (in-suspension breaks labeling in situ and sequencing) labels double-strand breaks (DSBs) in cells immobilized on glass coverslips, using double-stranded oligonucleotide adaptors that facilitate selective linear amplification through T7-mediated in vitro transcription (IVT), followed by next-generation sequencing (NGS) library preparation
|
No |
References
Ager, D. D., et al. (1990). Measurement of radiation-induced DNA double-strand breaks by pulsed-field gel electrophoresis. Radiation research, 122/(2), 181–187.
Anderson, D. & Laubenthal J. (2013), “Analysis of DNA Damage via Single-Cell Electrophoresis. In: Makovets S, editor. DNA Electrophoresis. Totowa.”, NJ: Humana Press. p 209-218.
Asaithamby, A., B. Hu and D.J. Chen. (2011) “Unrepaired clustered DNA lesions induce chromosome breakage in human cells.” Proc Natl Acad Sci U S A 108(20): 8293-8298 .
Barbieri, S., G. Babini, J. Morini et a l (2019). . Predicting DNA damage foci and their experimental readout with 2D microscopy: a unified approach applied to photon and neutron exposures. Scientific Reports 9(1): 14019
Bouwman, B. et al. (2020), “Genome-wide detection of DNA double-strand breaks by in-suspension BLISS”, Nature protocols,.15/12, Springer Nature, London, https://doi.org/10.1038/s41596-020-0397-2
Bryce, S. et al. (2016), “Genotoxic mode of action predictions from a multiplexed flow cytometric assay and a machine learning approach.”, Environ Mol Mutagen. 57:171-189. Doi: 10.1002/em.21996.
Burma, S. et al. (2001), “ATM phosphorylates histone H2AX in response to DNA double-strand breaks.”, J Biol Chem, 276(45): 42462-42467. doi:10.1074/jbc.C100466200
Cannan, W.J. and D.S. Pederson (2016), "Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin.", Journal of Cellular Physiology, Vol.231(/1), Wiley, New York, https://doi.org/10.1002/jcp.25048.
Cencer, C. et al. (2018), “PARP-1/PAR Activity in Cultured Human Lens Epithelial Cells Exposed to Two Levels of UVB Light”, Photochemistry and Photobiology, Vol.(94/1), Wiley-Blackwell, Hoboken, https://doi.org/10.1111/php.12814.
Charlton, E. D. et al. (1989), “Calculation of Initial Yields of Single and Double Stranded Breaks in Cell Nuclei from Electrons, Protons, and Alpha Particles.”, Int. J. Radiat. Biol. 56(1): 1-19. doi: 10.1080/09553008914551141.
Collins, R. A. (2004), “The Comet Assay for DNA Damage and Repair. Molecular Biotechnology.”, Mol Biotechnol. 26(3): 249-61. doi:10.1385/MB:26:3:249
EPRI (2014), Epidemiology and mechanistic effects of radiation on the lens of the eye: Review and scientific appraisal of the literature, EPRI, California.
Figueroa‑González, G. and C. Pérez‑Plasencia. (2017), “Strategies for the evaluation of DNA damage and repair mechanisms in cancer”, Oncology Letters, Vol.133(/6), Spandidos Publications, Athens, https://doi.org/10.3892/ol.2017.6002.
Garcia-Canton, C. et al. (2013), “Assessment of the in vitro p-H2AX assay by High Content Screening asa novel genotoxicity test.”, Mutat Res. 757:158-166. Doi: 10.1016/j.mrgentox.2013.08.002
Gardiner, K. et al. (1986), “Fractionation of Large Mammalian DNA Restriction Fragments Using Vertical Pulsed-Field Gradient Gel Electrophoresis.”, Somatic Cell and Molecular Genetics. 12(2): 185-95.Doi: 10.1007/bf01560665.
Garm, C. et al. (2012), “Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells”, Aging Cell, Vol.12/1, Blackwell Publishing Ltd, Oxford, https://doi.org/10.1111/acel.12019.
Hamada, N. (2014), “What are the intracellular targets and intratissue target cells for radiation effects?”, Radiation research, Vol. 181/1, The Radiation Research Society, Indianapolis, https://doi.org/10.1667/RR13505.1.
Herschleb, J. et al. (2007), “Pulsed-field gel electrophoresis.”, Nat Protoc. 2(3): 677-684. doi:10.1038/nprot.2007.94
Iliakis, G. et al. (2015), “Alternative End-Joining Repair Pathways Are the Ultimate Backup for Abrogated Classical Non-Homologous End-Joining and Homologous Recombination Repair: Implications for the Formation of Chromosome Translocations.”, Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2(3): 677-84. doi: 10.1038/nprot.2007.94
Jackson, S. (2002). “Sensing and repairing DNA double-strand breaks.”, Carcinogenesis. 23:687-696. Doi:10.1093/carcin/23.5.687.
Ji, J. et al. (2017), “Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay.”, PLoS One. 12(2): e0171582. doi:10.1371/journal.pone.0171582
Kawashima, Y.(2017), “Detection of DNA double-strand breaks by pulsed-field gel electrophoresis.”, Genes Cells 22:84-93. Doi: 10.1111/gtc.12457.
Khoury, L. et al. (2013), “Validation of high-throughput genotoxicity assay screening using cH2AX in-cell Western assay on HepG2 cells.”, Environ Mol Mutagen, 54:737-746. Doi: 10.1002/em.21817.
Khoury, L. et al. (2016), “Evaluation of four human cell lines with distinct biotransformation properties for genotoxic screening.”, Mutagenesis, 31:83-96. Doi: 10.1093/mutage/gev058.
Kohn, K.W. (1991), “Principles and practice of DNA filter elution”, Pharmacology & Therapeutics, Vol.49(/1), Elsevier, Amsterdam, https://doi.org/10.1016/0163-7258(91)90022-E.
Loo, DT. (2011), “In Situ Detection of Apoptosis by the TUNEL Assay: An Overview of Techniques. In: Didenko V, editor. DNA Damage Detection In Situ, Ex Vivo, and In Vivo. Totowa.”, NJ: Humana Press. p 3-13.doi: 10.1007/978-1-60327-409-8_1.
Mah, L. J. et al. (2010), “Quantification of gammaH2AX foci in response to ionising radiation.”, J Vis Exp(38). doi:10.3791/1957.
Nacci, D. et al. (1992), “Application of the DNA alkaline unwinding assay to detect DNA strand breaks in marine bivalves”, Marine Environmental Research, Vol.33(/2), Elsevier BV, Amsterdam, https://doi.org/10.1016/0141-1136(92)90134-8.
Nikolova, T., F. et al. (2017), “Genotoxicity testing: Comparison of the γH2AX focus assay with the alkaline and neutral comet assays.”, Mutat Res 822:10-18. Doi: 10.1016/j.mrgentox.2017.07.004.
Nitiss, J. L. et al. (2012), “Topoisomerase assays. ”, Curr Protoc Pharmacol. Chapter 3: Unit 3 3.
OECD. (2014). Test No. 489: “In vivo mammalian alkaline comet assay.” OECD Guideline for the Testing of Chemicals, Section 4 .
Olive, P. L., & Banáth, J. P. (2006), “The comet assay: a method to measure DNA damage in individual cells.”, Nature Protocols. 1(1): 23-29. doi:10.1038/nprot.2006.5.
Platel A. et al. (2011), “Study of oxidative DNA damage in TK6 human lymphoblastoid cells by use of the thymidine kinase gene-mutation assay and the in vitro modified comet assay: Determination of No-Observed-Genotoxic-Effect-Levels.”, Mutat Res 726:151-159. Doi: 10.1016/j.mrgentox.2011.09.003.
Raschke, S., J. Guan and G. Iliakis. (2009), “Application of alkaline sucrose gradient centrifugation in the analysis of DNA replication after DNA damage”, Methods in Molecular Biology, Vol.521, Humana Press, Totowa, https://doi.org/10.1007/978-1-60327-815-7_18.
Redon, C. et al. (2010), “The use of gamma-H2AX as a biodosimeter for total-body radiation exposure in non-human primates.”, PLoS One. 5(11): e15544. doi:10.1371/journal.pone.0015544
Revet, I. et al. (2011), “Functional relevance of the histone γH2Ax in the response to DNA damaging agents.” Proc Natl Acad Sci USA.108:8663-8667. Doi: 10.1073/pnas.1105866108
Rogakou, E.P. et al. (1998), “DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139.” , J Biol Chem, 273:5858-5868. Doi: 10.1074/jbc.273.10.5858
Rothkamm, K. & Horn, S. (2009), “γ-H2AX as protein biomarker for radiation exposure.”, Ann Ist Super Sanità, 45(3): 265-71.
White, R.R. and J. Vijg. (2016), “Do DNA Double-Strand Breaks Drive Aging?”, Molecular Cell, Vol.63, Elsevier, Amsterdam, http://doi.org/10.1016/j.molcel.2016.08.004.
Yang, Y. et al. (1998), “The effect of catalase amplification on immortal lens epithelial cell lines”, Experimental Eye Research, Vol.67(/6), Academic Press Inc, Cambridge, https://doi.org/10.1006/exer.1998.0560.
Zilio, N. and H. D. Ulrich (2021), “Exploring the SSBreakome: genome-wide mapping of DNA single-strand breaks by next-generation sequencing”, The FEBS journal, 288(13), Wiley, Hoboken, https://doi.org/10.1111/febs.15568
Event: 1262: Apoptosis
Short Name: Apoptosis
Event Component
| Process | Object | Action |
|---|---|---|
| apoptotic process | increased |
AOPs Including This Key Event
Biological Context
| Level of Biological Organization |
|---|
| Cellular |
Cell term
| Cell term |
|---|
| cell |
Organ term
| Organ term |
|---|
| organ |
Domain of Applicability
Taxonomic Applicability| Term | Scientific Term | Evidence | Links |
|---|---|---|---|
| Homo sapiens | Homo sapiens | High | NCBI |
| Mus musculus | Mus musculus | High | NCBI |
| Rattus norvegicus | Rattus norvegicus | High | NCBI |
| Caenorhabditis elegans | Caenorhabditis elegans | High | NCBI |
| Life Stage | Evidence |
|---|---|
| Not Otherwise Specified | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
・Apoptosis is induced in human prostate cancer cell lines (Homo sapiens) [Parajuli et al., 2014].
・Apoptosis occurs in B6C3F1 mouse (Mus musculus) [Elmore, 2007].
・Apoptosis occurs in Sprague-Dawley rat (Rattus norvegicus) [Elmore, 2007].
・Apoptosis occurs in the nematode (Caenorhabditis elegans) [Elmore, 2007].
- Apoptosis occurs in breast cancer cells, human and mouse (Parton)
- Apoptosis applicable to fishes, hence be used to study as models (dos Santos, N. M., et al. (2008).
- Apoptosis in humans and baboon ovaries (Kugu, K., et al. (1998)
- Apoptosis in amphibians during metamorphosis (Ishizuya-Oka, A., et al. (2010).
- Apoptosis in Drosophila melanogaster (Steller, H. (2008)
- Apoptosis is a highly conserved and essential process across a broad taxonomic range, from unicellular eukaryotes to complex multicellular animals, it is also evident in metazoans (Suraweera, C. D., et al. (2022).
-
Sex Applicability:
Both sexes. Apoptosis occurs in male and female systems (e.g., oocyte and sperm cell turnover). -
Life Stage Applicability:
All stages. Especially critical during embryonic development and in maintaining adult tissue homeostasis.
Key Event Description
Apoptosis, the process of programmed cell death, is characterized by distinct morphology with DNA fragmentation and energy dependency [Elmore, 2007]. Apoptosis, also called “physiological cell death”, is involved in cell turnover, physiological involution, and atrophy of various tissues and organs [Kerr et al., 1972]. The formation of apoptotic bodies involves marked condensation of both nucleus and cytoplasm, nuclear fragmentation, and separation of protuberances [Kerr et al., 1972]. Apoptosis is characterized by DNA ladder and chromatin condensation. Several stimuli such as hypoxia, nucleotides deprivation, chemotherapeutical drugs, DNA damage, and mitotic spindle damage induce p53 activation, leading to p21 activation and cell cycle arrest [Pucci et al., 2000]. The SAHA or TSA treatment on neonatal human dermal fibroblasts (NHDFs) for 24 or 72 hrs inhibited proliferation of the NHDF cells [Glaser et al., 2003]. Considering that the acetylation of histone H4 was increased by the treatment of SAHA for 4 hrs, histone deacetylase inhibition may be involved in the inhibition of the cell proliferation [Glaser et al., 2003]. The impaired proliferation was observed in HDAC1-/- ES cells, which was rescued with the reintroduction of HDAC1 [Zupkovitz et al., 2010]. An AOP focuses existes on p21 pathway leading to apoptosis, however, alternative pathways such as NF-kappaB signaling pathways may be involved in the apoptosis of spermatocytes [Wang et al., 2017].
Apoptosis is defined as a programmed cell death. A decrease in apoptosis or a resistance to cell death is noted is described as a hallmark of cancer by Hanahan et al. It is widely admitted as an essential step in tumor proliferation (Adams, Lowe). Apoptosis occurs after activation of a number of intrinsic and extrinsic signals which activate the protease caspase system which in turn activates the destruction of the cell.
In mammals, the foetal ovary produces hundreds of thousands of oocytes. But most of them die before birth due to apoptosis (Kaur, S., & Kurokawa, M., 2023). The apoptotic process has a specific pattern at different stages: in foetal ovaries, the majority of apoptotic activity was found in germ cells, whereas in adult quiescent cortical follicles, apoptosis occurred from both granulosa and oocyte cells. The oocyte has been shown to be the one that triggers the apoptotic process and causes follicular atresia (Jin, X., et al. (2011). In humans, the primordial follicles' ovarian endowment is formed throughout foetal development. Apoptotic cell death, which is carried out with the assistance of multiple players and routes conserved from worms to humans, depletes this endowment by at least two-thirds prior to birth. As of right now, apoptosis has been linked to atresia, oocyte loss/selection, folliculogenesis, and oogenesis (Hussein MR, 2005)
The Bcl-2 is a protein family suppressing apoptosis by binding and inhibiting two proapoptotic proteins (Bax and Bak) and transferring them to the mitochondrial outer membrane. In the absence of inhibition by Bcl2, Bax and Bak destroy the mitochondrial membrane and releases proapoptotic signaling proteins, such as cytochrome c which activated the caspase system. An increased expression of these antiapoptotic proteins (Bcl-2, Bcl-xL) occurs in cancer (Hanahan, Adams, Lowe). Several others pathways such as the loss of TP53 tumor suppressor function, or the increase of survival signals (Igf1/2), or decrease of proapoptotic factors (Bax, Bim, Puma) can also increase tumor growth (Hanahan, Juntilla).
In breast cancer a decrease in apoptosis and a resistance to cell death has been described thoroughly, especially using a dysregulation of the Bcl2 system or TP53 (Parton, Williams, Shahbandi).
How it is Measured or Detected
Apoptosis is characterized by many morphological and biochemical changes such as homogenous condensation of chromatin to one side or the periphery of the nuclei, membrane blebbing and formation of apoptotic bodies with fragmented nuclei, DNA fragmentation, enzymatic activation of pro-caspases, or phosphatidylserine translocation that can be measured using electron and cytochemical optical microscopy, proteomic and genomic methods, and spectroscopic techniques [Archana et al., 2013; Martinez et al., 2010; Taatjes et al., 2008; Yasuhara et al., 2003].
・DNA fragmentation can be quantified with comet assay using electrophoresis, where the tail length, head size, tail intensity, and head intensity of the comet are measured [Yasuhara et al., 2003].
・The apoptosis is detected with the expression alteration of procaspases 7 and 3 by Western blotting using antibodies [Parajuli et al., 2014].
・The apoptosis is measured with down-regulation of anti-apoptotic gene baculoviral inhibitor of apoptosis protein repeat containing 2 (BIRC2, or cIAP1) [Parajuli et al., 2014].
・Apoptotic nucleosomes are detected using Cell Death Detection ELISA kit, which was calculated as absorbance subtraction at 405 nm and 490 nm [Parajuli et al., 2014].
・Cleavage of PARP is detected with Western blotting [Parajuli et al., 2014].
・Caspase-3 and caspase-9 activity is measured with the enzyme-catalyzed release of p-nitroanilide (pNA) and quantified at 405 nm [Wu et al., 2016].
・Apoptosis is measured with Annexin V-FITC probes, and the relative percentage of Annexin V-FITC-positive/PI-negative cells is analyzed by flow cytometry [Wu et al., 2016].
・Apoptosis is detected with the Terminal dUTP Nick End-Labeling (TUNEL) method to assay the endonuclease cleavage products by enzymatically end-labeling the DNA strand breaks [Kressel and Groscurth, 1994].
・For the detection of apoptosis, the testes are fixed in neutral buffered formalin and embedded in paraffin. Germ cell death is visualized in testis sections by Terminal dUTP Nick End-Labeling (TUNEL) staining method [Wade et al., 2008]. The incidence of TUNEL-positive cells is expressed as the number of positive cells per tubule examined for one entire testis section per animal [Wade et al., 2008]
References
Archana, M. et al. (2013), "Various methods available for detection of apoptotic cells", Indian J Cancer 50:274-283
Elmore, S. (2007), "Apoptosis: a review of programmed cell death", Toxicol Pathol 35:495-516
Glaser, K.B. et al. (2003), "Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines", Mol Cancer Ther 2:151-163
Kerr, J.F.R. et al. (1972), "Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics", Br J Cancer 26:239-257
Kressel, M. and Groscurth, P. (1994), "Distinction of apoptotic and necrotic cell death by in situ labelling of fragmented DNA", Cell Tissue Res 278:549-556
Martinez, M.M. et al. (2010), "Detection of apoptosis: A review of conventioinal and novel techniques", Anal Methods 2:996-1004
Parajuli, K.R. et al. (2014), "Methoxyacetic acid suppresses prostate cancer cell growth by inducing growth arrest and apoptosis", Am J Clin Exp Urol 2:300-313
Pucci, B. et al. (2000), "Cell cycle and apoptosis", Neoplasia 2:291-299
Taatjes, D.J. et al. (2008), "Morphological and cytochemical determination of cell death by apoptosis", Histochem Cell Biol 129:33-43
Wade, M.G. et al. (2008), "Methoxyacetic acid-induced spermatocyte death is associated with histone hyperacetylation in rats", Biol Reprod 78:822-831
Wang, C. et al. (2017), "CD147 regulates extrinsic apoptosis in spermatocytes by modulating NFkB signaling pathways", Oncotarget 8:3132-3143
Wu, R. et al. (2016), "microRNA-497 induces apoptosis and suppressed proliferation via the Bcl-2/Bax-caspase9-caspase 3 pathway and cyclin D2 protein in HUVECs", PLoS One 11:e0167052
Yasuhara, S. et al. (2003), "Comparison of comet assay, electron microscopy, and flow cytometry for detection of apoptosis", J Histochem Cytochem 51:873-885
Zupkovitz, G. et al. (2010), "The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation", Mol Cell Biol 30:1171-1181
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230
Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007 Feb 26;26(9):1324-37. doi: 10.1038/sj.onc.1210220. PMID: 17322918; PMCID: PMC2930981.
Lowe, S., Cepero, E. & Evan, G. Intrinsic tumour suppression. Nature 432, 307–315 (2004). https://doi.org/10.1038/nature03098
Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001 Jun 23;322(7301):1528-32. doi: 10.1136/bmj.322.7301.1528. PMID: 11420276; PMCID: PMC1120573.
Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009 Nov;9(11):821-9. doi: 10.1038/nrc2728. Epub 2009 Sep 24. PMID: 19776747.
Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015 Feb 28;6(6):3519-30. doi: 10.18632/oncotarget.2792. PMID: 25784482; PMCID: PMC4414133.
Shahbandi A, Nguyen HD, Jackson JG. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer. 2020 Feb;6(2):98-110. doi: 10.1016/j.trecan.2020.01.007. Epub 2020 Feb 5. PMID: 32061310; PMCID: PMC7931175.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230
Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007 Feb 26;26(9):1324-37. doi: 10.1038/sj.onc.1210220. PMID: 17322918; PMCID: PMC2930981.
Lowe, S., Cepero, E. & Evan, G. Intrinsic tumour suppression. Nature 432, 307–315 (2004). https://doi.org/10.1038/nature03098
Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001 Jun 23;322(7301):1528-32. doi: 10.1136/bmj.322.7301.1528. PMID: 11420276; PMCID: PMC1120573.
Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009 Nov;9(11):821-9. doi: 10.1038/nrc2728. Epub 2009 Sep 24. PMID: 19776747.
Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015 Feb 28;6(6):3519-30. doi: 10.18632/oncotarget.2792. PMID: 25784482; PMCID: PMC4414133.
Shahbandi A, Nguyen HD, Jackson JG. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer. 2020 Feb;6(2):98-110. doi: 10.1016/j.trecan.2020.01.007. Epub 2020 Feb 5. PMID: 32061310; PMCID: PMC7931175.
Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001 Jun 23;322(7301):1528-32. doi: 10.1136/bmj.322.7301.1528. PMID: 11420276; PMCID: PMC1120573.
- Kaur S, Kurokawa M. Regulation of Oocyte Apoptosis: A View from Gene Knockout Mice. Int J Mol Sci. 2023;24(2).
- Jin X, Xiao LJ, Zhang XS, Liu YX. Apotosis in ovary. Front Biosci (Schol Ed). 2011;3(2):680-97.
- Hussein MR. Apoptosis in the ovary: molecular mechanisms. Hum Reprod Update. 2005;11(2):162-77.
- dos Santos NM, do Vale A, Reis MI, Silva MT. Fish and apoptosis: molecules and pathways. Curr Pharm Des. 2008;14(2):148-69.
- Kugu K, Ratts VS, Piquette GN, Tilly KI, Tao XJ, Martimbeau S, et al. Analysis of apoptosis and expression of bcl-2 gene family members in the human and baboon ovary. Cell Death Differ. 1998;5(1):67-76.
- Ishizuya-Oka A, Hasebe T, Shi YB. Apoptosis in amphibian organs during metamorphosis. Apoptosis. 2010;15(3):350-64.
- Steller H. Regulation of apoptosis in Drosophila. Cell Death & Differentiation. 2008;15(7):1132-8.
- Suraweera CD, Banjara S, Hinds MG, Kvansakul M. Metazoans and Intrinsic Apoptosis: An Evolutionary Analysis of the Bcl-2 Family. International Journal of Molecular Sciences. 2022;23(7):3691.
List of Adverse Outcomes in this AOP
Event: 1757: Decrease, Sperm count
Short Name: Decrease, Sperm count
AOPs Including This Key Event
| AOP ID and Name | Event Type |
|---|---|
| Aop:322 - Alkylation of DNA leading to decreased sperm count | AdverseOutcome |
| Aop:400 - Inhibition of CYP26B1 activity in fetal testis leading to reduced fertility | KeyEvent |
| Aop:616 - organic UV filter and its Photoproducts reproductive toxicity pathways | KeyEvent |
Biological Context
| Level of Biological Organization |
|---|
| Individual |
Domain of Applicability
This KE is plausibly applicable to all male animals that produce sperm through spermatogenesis.
Key Event Description
Sperm is produced in the seminiferous tubules of the testis through spermatogenesis (Sharma & Agarwal, 2011). This process begins with Type A spermatogonia, which divide by mitosis to maintain the stem cell pool. A subset of these cells differentiate into Type B spermatogonia, which divide by mitosis and give rise to primary spermatocytes. Subsequently, primary spermatocytes undergo meiosis I to form secondary spermatocytes, which undergo meiosis II to produce haploid spermatids. Spermatids then differentiate into spermatozoa through spermiogenesis, a process marked by several morphological changes, including condensation and elongation of the nucleus, acrosome formation, and development of the flagellum (Nishimura & L’Hernault, 2017; Sharma & Agarwal, 2011). Spermatozoa are released into the seminiferous tubule lumen, exit the testis through the rete testis, and enter the epididymis, where sperm maturation occurs. Spermatozoa acquire motility and acrosomal function during transit through the three distinct regions of the epididymis: caput, corpus, and cauda (Sharma & Agarwal, 2011).
Sperm count refers to the number of spermatids present in the testis, or the number of spermatozoa present in semen or the cauda epididymis. Reduced sperm count describes a decrease in spermatids or spermatozoa with respect to a control or reference number. In humans, a total sperm number below 39 million per ejaculate and a sperm concentration below 16 million per ml represent the fifth percentile lower limits, based on a reference group of men whose partners conceived within 12 months (World Health Organization, 2021). Reduced sperm count can be temporary, prolonged, or permanent depending on the cause, including genetic or other intrinsic problems, or an exposure that occurred during development that impaired the stem cell pool.
The toxicological interpretation of reduced sperm count depends on the biological compartment assessed. A decrease in testicular spermatid number suggests impairment of one or more stages of spermatogenesis (Creasy & Chapin, 2013; M. L. Meistrich, 1989). However, a reduction in cauda epididymal sperm reserves may reflect impaired spermatogenesis or spermatid retention in the testis, disruption of epididymal processes such as sperm transit, maturation, and storage, or a combination of these effects (Blazak et al., 1985; Creasy & Chapin, 2013).
The timing and duration of exposure are important considerations when evaluating sperm count due to the length of spermatogenesis and the spermatogenic cycle. Exposure durations spanning multiple spermatogenic cycles may be necessary to produce detectable changes in sperm count, as toxicants that target earlier stages of spermatogenesis may only affect sperm count after the damaged cells have progressed through subsequent stages of development (Amann, 1986; Mangelsdorf et al., 2003). For epididymal sperm counts, sperm transit time through the epididymis should also be considered.
How it is Measured or Detected
OECD Test Guideline 416: Two-Generation Reproduction Toxicity, and OECD Test Guideline 443: Extended One-Generation Reproductive Toxicity Study, recommend estimating sperm count by quantifying cauda epididymis sperm reserves and spermatids in the testis (OECD, 2001, 2025).
Sperm counts can be estimated from the testis, epididymis, or semen. Testicular sperm counts are generally estimated by quantifying homogenization-resistant spermatids (Amann, 1986). During spermiogenesis, spermatid nuclei become highly condensed and resistant to mechanical or biochemical breakdown. Homogenization destroys most testicular cells and nuclei except for the late-stage spermatid nuclei, which can then be quantified (Amann, 1986). Testicular sperm counts can also be used to estimate daily sperm production (DSP) by dividing the number of homogenization-resistant spermatid nuclei by the number of days they spend in the testis (Amann, 1981).
Epididymal sperm counts are most often estimated using the cauda epididymis, where sperm is stored (Seed et al., 1996). Sperm can be isolated from the cauda epididymis using various methods, including diffusion, aspiration, or homogenization (Chapin et al., 1992; Seed et al., 1996; Slott et al., 1991). In the diffusion method, small incisions are made in the cauda epididymis to allow sperm to swim out into the surrounding medium. The aspiration method collects sperm directly from incised tissue using a capillary tube. Homogenization methods mechanically disrupt epididymal tissue to release sperm (Amann, 1986; Seed et al., 1996).
In species where semen can be collected, such as humans, dogs, and rabbits, sperm count can be evaluated from ejaculated semen samples (Seed et al., 1996).
The resulting sperm suspension is counted manually or by automated methods. Manual counting using a hemacytometer and phase-contrast microscopy is a widely used and accepted method for determining sperm count (Amann, 1986; Seed et al., 1996; Strader et al., 1996). Sperm counting with a hemacytometer, specifically the improved Neubauer hemacytometer, is considered the gold standard and is extensively described in the WHO laboratory manual for the examination and processing of human semen (World Health Organization, 2021). Hemacytometer counts are used for calibrating other automated techniques (Kuster, 2005; Prathalingam et al., 2006).
Automated methods include Computer-Assisted Sperm Analysis (CASA), in which a video camera attached to a microscope captures images or videos that are analyzed by specialized software (Akal, 2023). The CASA system objectively estimates sperm concentration and related sperm parameters. CASA-derived sperm counts have demonstrated strong agreement with hemacytometer-based methods while improving analytical efficiency (Dearing et al., 2014; Lammers et al., 2014; Strader et al., 1996). However, CASA systems have been reported to overestimate sperm count at lower concentrations due to misclassification of debris as sperm (Dearing et al., 2014).
Flow cytometry-based approaches have also been developed for counting sperm and assessing sperm membrane integrity. Sperm from zebrafish testis were stained with SYBR-14, a membrane-permeable nucleic acid dye, and propidium iodide, a DNA dye that can only permeate damaged cell membranes. Fluorescence filters were used to detect stained cells, and forward scatter (FSC) and side scatter (SSC) were used to differentiate sperm from debris. Resulting sperm counts were comparable to those obtained from using a hemacytometer (Yang et al., 2016).
References
Akal, E. (2023). Evaluation of sperm counting accuracy on computer-assisted sperm analysis with GoldCyto® slides and glass slides. Frontiers in Veterinary Science, 10, 1283128. https://doi.org/10.3389/fvets.2023.1283128
Amann, R. P. (1981). A Critical Review of Methods for Evaluation of Spermatogenesis from Seminal Characteristics. Journal of Andrology, 2(1), 37–58. https://doi.org/10.1002/j.1939-4640.1981.tb00595.x
Amann, R. P. (1986). Detection of alterations in testicular and epididymal function in laboratory animals. Environmental Health Perspectives, 70, 149–158. https://doi.org/10.1289/ehp.8670149
Blazak, W. F., Ernst, T. L., & Stewart, B. E. (1985). Potential indicators of reproductive toxicity: Testicular sperm production and epididymal sperm number, transit time, and motility in Fischer 344 rats. Fundamental and Applied Toxicology, 5(6, Part 1), 1097–1103. https://doi.org/10.1016/0272-0590(85)90145-9
Chapin, R. E., Filler, R. S., Gulati, D., Heindel, J. J., Katz, D. F., Mebus, C. A., Obasaju, F., Perreault, S. D., Russell, S. R., & Schrader, S. (1992). Methods for assessing rat sperm motility. Reproductive Toxicology, 6(3), 267–273. https://doi.org/10.1016/0890-6238(92)90183-t
Creasy, D. M., & Chapin, R. E. (2013). Male Reproductive System. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology (pp. 2493–2598). Academic Press. https://doi.org/10.1016/B978-0-12-415759-0.00059-5
Dearing, C. G., Kilburn, S., & Lindsay, K. S. (2014). Validation of the sperm class analyser CASA system for sperm counting in a busy diagnostic semen analysis laboratory. Human Fertility, 17(1), 37–44. https://doi.org/10.3109/14647273.2013.865843
Kuster, C. (2005). Sperm concentration determination between hemacytometric and CASA systems: Why they can be different. Theriogenology, Proceedings of the 2005 Annual Conference of the Society for Theriogenology, 64(3), 614–617. https://doi.org/10.1016/j.theriogenology.2005.05.047
Lammers, J., Splingart, C., Barrière, P., Jean, M., & Fréour, T. (2014). Double-blind prospective study comparing two automated sperm analyzers versus manual semen assessment. Journal of Assisted Reproduction and Genetics, 31(1), 35–43. https://doi.org/10.1007/s10815-013-0139-2
M. L. Meistrich. (1989). Evaluation of Reproductive Toxicity by Testicular Sperm Head Counts. 8(3), 551–567. https://doi.org/10.3109/10915818909014538
Mangelsdorf, I., Buschmann, J., & Orthen, B. (2003). Some aspects relating to the evaluation of the effects of chemicals on male fertility. Regulatory Toxicology and Pharmacology, 37(3), 356–369. https://doi.org/10.1016/S0273-2300(03)00026-6
OECD. (2001). Test No. 416: Two-Generation Reproduction Toxicity. OECD. https://doi.org/10.1787/9789264070868-en
OECD. (2025). Test No. 443: Extended One-Generation Reproductive Toxicity Study. OECD Publishing. https://doi.org/10.1787/9789264185371-en
Prathalingam, N. S., Holt, W. W., Revell, S. G., Jones, S., & Watson, P. F. (2006). The Precision and Accuracy of Six Different Methods to Determine Sperm Concentration. Journal of Andrology, 27(2), 257–262. https://doi.org/10.2164/jandrol.05112
Seed, J., Chapin, R. E., Clegg, E. D., Dostal, L. A., Foote, R. H., Hurtt, M. E., Klinefelter, G. R., Makris, S. L., Perreault, S. D., Schrader, S., Seyler, D., Sprando, R., Treinen, K. A., Veeramachaneni, D. N. R., & Wise, L. D. (1996). Methods for assessing sperm motility, morphology, and counts in the rat, rabbit, and dog: A consensus report. Reproductive Toxicology, 10(3), 237–244. https://doi.org/10.1016/0890-6238(96)00028-7
Sharma, R., & Agarwal, A. (2011). Spermatogenesis: An Overview. In A. Zini & A. Agarwal (Eds.), Sperm Chromatin: Biological and Clinical Applications in Male Infertility and Assisted Reproduction (pp. 19–44). Springer. https://doi.org/10.1007/978-1-4419-6857-9_2
Slott, V. L., Suarez, J. D., & Perreault, S. D. (1991). Rat sperm motility analysis: Methodologic considerations. Reproductive Toxicology, 5(5), 449–458. https://doi.org/10.1016/0890-6238(91)90009-5
Strader, L. F., Linder, R. E., & Perreault, S. D. (1996). Comparison of rat epididymal sperm counts by IVOS HTM-IDENT and hemacytometer. Reproductive Toxicology, 10(6), 529–533. https://doi.org/10.1016/S0890-6238(96)00140-2
World Health Organization. (2021). WHO Laboratory Manual for the Examination and Processing of Human Semen (6th ed). WHO Press.
Yang, H., Daly, J., & Tiersch, T. R. (2016). Determination of Sperm Concentration Using Flow Cytometry with Simultaneous Analysis of Sperm Plasma Membrane Integrity in Zebrafish Danio rerio. Cytometry. Part A : The Journal of the International Society for Analytical Cytology, 89(4), 350–356. https://doi.org/10.1002/cyto.a.22796
Appendix 2
List of Key Event Relationships in the AOP
List of Adjacent Key Event Relationships
Relationship: 24: Alkylation, DNA leads to Inadequate DNA repair
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Alkylation of DNA in male pre-meiotic germ cells leading to heritable mutations | adjacent | High | Moderate |
| Alkylation of DNA leading to cancer 1 | adjacent | High | Moderate |
| Alkylation of DNA leading to decreased sperm count | adjacent | High | Moderate |
Evidence Supporting Applicability of this Relationship
DNA adducts can occur in any cell type. While there are differences across taxa, all species have some DNA repair systems in place and it is common to extrapolate conclusions across eukaryotic species.
Key Event Relationship Description
Alkylated DNA may be tolerated and/or repaired error-free by a variety of DNA repair pathways. However, at high doses, it is established that the primary DNA repair pathway (O6-Alkylguanine-DNA alkyltransferase: AGT) responsible for removing alkylated DNA becomes saturated. This may lead to several potential adduct fates: (i) error-free repair of the DNA adduct using alternative DNA repair mechanisms; (ii) no repair (DNA damage is retained); or (iii) instability in the DNA duplex leading to DNA strand breaks and possibly activation of DNA damage signaling. For repair of alkyl adducts it is well established that the O6-alkylguanine-DNA alkyltransferase pathway becomes saturated at high doses leading to insufficient repair at high doses.
Evidence Supporting this KER
Biological PlausibilityGeneral details: The weight of evidence for this KER is strong. It is widely accepted that damaged DNA is subject to repair, and that in the absence of DNA repair, mutations will ensue. Specifically, AGT (Damage Reversal DNA repair: pathway #1 in KE155), also known as O6-methylguanine-DNA methyltransferase (MGMT), reverses alkylation damage by directly transferring alkyl groups from the O6 position of guanine to a cysteine residue on the AGT (or MGMT) molecule, restoring the DNA in a single step. However, transfer of the alky group to AGT results in concomitant inactivation of AGT (Pegg 2011). The mammalian protein is also active on O6-ethylguanine and can remove only one ethyl group from DNA, following which the protein is degraded. Thus, high levels of alkylation damage overwhelm the cellular AGT capacity to remove lesions. In mammalian cells, O4-ethylthymine and O2-ethylthymine are poor substrates for AGT (Fang et al. 2010) and no other DNA repair pathway has been identified that is able to efficiently repair these lesions; consequently, these lesions are extremely persistent in cells. Reviews on this topic have been published (Kaina et al. 2007; Pegg 2011). In the absence of the AGT/MGMT pathway, other DNA repair pathways may be invoked, but the relative efficiency of these pathways is not well understood (further details described below).
The role of nucleotide excision repair (NER; excision repair pathways: #2 in KE155) in alkylation damage repair in mammalian cells remains unclear. Earlier studies using human cell lines suggested that both AGT and NER may be involved in the repair of O6-ethylguanine (Bronstein et al. 1991; Bronstein et al. 1992). Very recently, an alkyltransferase like protein (ATL1) that has homology to AGT has been identified in a range of prokaryotes and lower eukaryotes. This protein has no alkyltransferase activity but can couple O6-alkylguanine damage to NER (Latypov et al. 2012). ATL1 proteins have not yet been identified in mammals.
Some alkyl adducts, such as N7-ethylguanine and N3-ethyladenine, are inherently unstable and may depurinate (i.e., hydrolytic cleavage of the glycosidic bond, which releases adenine or guanine). The resultant abasic sites are normally repaired through error-free pathways although they may occasionally be transformed to DNA strand breaks. In mammals, N-methylpurine DNA glycosylases, such as alkyladenine DNA glycosylase (AAG), have a wide range of substrates including N7-alkylguanine and N3-alkyladenine derivatives (Wyatt et al. 1999). However, there are no specific reports in the literature that the ethylated derivatives are AAG substrates. Glycosylases such as AAG yield abasic sites that are processed as described above. An alternative repair mechanism for repairing minor lesions such as N3-ethylcytosine and N1-ethyladenine is through oxidative dealkylation catalyzed by AlkB and mammalian homologs (Drabløs et al. 2004). This pathway is an error-free damage reversal pathway that releases the oxidized ethyl group as acetaldehyde (Duncan et al. 2002).
A final mechanism through which DNA repair pathways may influence the fate of alkylation damage is through futile cycling of the mismatch repair (MMR; excision repair pathways: #2 in KE155) system at an O6-alkyl G:T mispair. In this scenario, unrepaired O6-alkylguanine is able to mispair with T, and the mispair is recognized by MMR enzymes resulting in the removal of the newly incorporated thymine from the nascent strand opposite the O6-alkyguanine adduct. During DNA repair synthesis, O6-alkylguanine preferentially pairs once again with thymine, reinitiating the repair/synthesis cycle. This iteration of excision and synthesis may produce strand breaks and activate damage signaling pathways (York and Modrich 2006).
If the pathways described above become saturated or do not operate properly, the alkylated DNA will not be repaired and will provide a template for replication of this damaged DNA. This is widely understood and accepted. Many studies have demonstrated that the introduction of plasmids or vectors with alkylated DNA (i.e., unrepaired lesions) into prokaryotic and eukaryotic cells, followed by replication, results in the formation of mutations at the alkylated sites, and that the probability of a mutation occurring at the alkylated site is modified by specific DNA repair genes/pathways (reviewed in Basu and Essigmann 1990; Shrivastav et al. 2010).
Empirical EvidenceInsufficient repair is inferred from the formation and retention of adducts, and the formation of increased numbers of mutations above background (i.e., KE185 - methodologies described therein).
A variety of studies show that alkylated DNA persists for prolonged periods of time post-exposure. For example, persistence of different alkylated nucleotides was shown in livers and brains of C57BL mice exposed to N-methyl-N-nitrosourea, N-ethyl-N-nitrosourea and ethyl methanesulfonate using high-performance liquid chromatography several days post-exposure (Frei et al., 1978). The stability of methyl and ethyl adducts in somatic tissues for various adduct types is summarized in Beranuk, 1990. The in vivo liver half life of methyl adducts ranges from 0-3 days, and liver ethyl adduct half lives can be up to 17 days, indicating poorer repair of oxygen-bound ethyl adducts. This prolonged retention of adducts indicates that there is insufficient repair by AGT or other DNA repair pathways of these adducts.
Studies in both hamsters and rats show persistence of alkylated nucleotides several days post-exposure, indicating lack of DNA repair of some adducts (Scherer et al. 1987; Seiler et al. 1997). For example, 101xC3H mouse hybrid testes exhibited DNA adducts within 1 hour of exposure to ENU (10 or 100 mg/kg by i.p.), but some adducts remained unrepaired six days post-exposure (Sega et al. 1986). O6-ethylguanine adducts were also found in hamster spermatogonia DNA up to four days after exposure to DEN (100 µg/g body weight) (Seiler et al. 1997). O6-ethylguanine adducts were found in spermatogonia 1.5 hours post-exposure to ENU in Syrian Golden hamsters (Seiler et al. 1997). Approximately 30% persisted in spermatogonia four days post-exposure. Moreover, the amount of O6-ethylguanine recovered after a 100 mg ENU/kg exposure was 40% greater than predicted from a linear extrapolation of the amount of O6-ethylguanine recovered after exposure to 10 mg/kg. The data suggest that the high dose exposure to ENU results in depletion of AGT within the testis and permits O6-ethylguanine to persist at higher levels than would be predicted from lower exposure. The relationship between dose and formation of DNA adducts in tubular germ cells is non-linear, indicating relatively rapid repair at low doses that becomes saturated at higher doses (van Zeeland et al. 1990). Thus, with increasing dose, increasing incidence of KE1 (insufficient repair) occurs. This implies that mouse spermatogonia are capable of repairing a major part of the DNA damage at low doses. However, at higher doses the repair process is saturated and mutations begin to occur. Indeed, the dose-response curve for mutations in spermatogonia measured in sperm of exposed males is sub-linear with a clear point of inflection at low sub-chronic doses of ENU (O’Brien et al. 2015).
Finally, both alkyl adducts and mutations increase with increasing doses of alkylating agents in somatic cells and in male germ cells, indicating that DNA repair processes are not operating to remove all of the damage (ability to remove adducts and prevent mutations).
Uncertainties and InconsistenciesDNA repair is not generally measured directly; thus, insufficient repair is inferred from the retention of adducts or the induction of increases in mutation frequencies post-exposure. In addition, various sizes of alkylation groups (e.g., methyl, ethyl, propyl) can be involved. Although it appears that the larger alkyl adducts tend to be more mutagenic (Beranek, 1990), this is not completely established and there are insufficient data to establish that this is true for germ cells. However, in general, this KER is biologically plausible, broadly accepted for alkyl adducts and has few uncertainties. The direct measurement of insufficient repair can be considered a data gap.
References
Basu, A.K. and J.M. Essigmann (1990), "Site-specific alkylated oligodeoxynucleotides: Probes for mutagenesis, DNA repair and the structure effects of DNA damage", Mutation Research, 233: 189-201.
Beranek, D.T. (1990), "Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents", Mutation Research , 231(1): 11-30.
Bronstein, S.M., J.E. Cochrane, T.R. Craft, J.A. Swenberg and T.R. Skopek (1991), "Toxicity, mutagenicity, and mutational spectra of N-ethyl-N-nitrosourea in human cell lines with different DNA repair phenotypes", Cancer Research, 51(19): 5188-5197.
Bronstein, S.M., T.R. Skopek and J.A. Swenberg (1992), "Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells", Cancer Research, 52(7): 2008-2011.
Drabløs, F., E. Feyzi, P.A. Aas, C.B. Vaagbø, B. Kavli, M.S. Bratlie, J. Peña-Diaz, M. Otterlei, G. Slupphaug and H.E. Krokan (2004), "Alkylation damage in DNA and RNA--repair mechanisms and medical significance", DNA Repair, 3(11): 1389-1407.
Duncan, T., S.C. Trewick, P. Koivisto, P.A. Bates, T. Lindahl and B. Sedgwick B (2002), "Reversal of DNA alkylation damage by two human dioxygenases", Proc. Natl. Acad. Sci. USA, 99(26): 16660-16665.
Fang, Q., S. Kanugula, J.L. Tubbs, T.A. Tainer and A.E. Pegg (2010), "Repair of O4-alkylthymine by O6-alkylguanine-DNA alkyltransferases", J. Biol. Chem. 12(285): 885-895.
Frei, J.V., D.H .Swenson, W. Warren, P.D. Lawley (1978), "Alkylation of deoxyribonucleic acid in vivo in various organs of C57BL mice by the carcinogens N-methyl-N-nitrosourea, N-ethyl-N-nitrosourea and ethyl methanesulphonate in relation to induction of thymic lymphoma. Some applications of high-pressure liquid chromatography", Biochem. J., 174(3): 1031-1044.
Kaina, B., M. Christmann, S. Naumann and W.P. Roos (2007), "MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents", DNA Repair, 6: 1079–1099.
Latypov, V.F., J.L. Tubbs, A.J. Watson, A.S. Marriott, G. McGown, M. Thorncroft, O.J. Wilkinson, P. Senthong, A. Butt, A.S. Arvai, C.L. Millington, A.C. Povey, D.M. Williams, M.F. Santibanez-Koref, J.A. Tainer and G.P. Margison GP (2012), "Atl1 regulates choice between global genome and transcription-coupled repair of O(6)-alkylguanines", Mol. Cell, 47(1): 50-60.
Muller, L., E. Gocke, T. Lave and T. Pfister (2009), "Ethyl methanesulfonate toxicity in Viracept – a comprehensive assessment based on threshold data for genotoxicity", Toxicology Letters, 190: 317-329.
O’Brien, J.M., M. Walker, A. Sivathayalan, G.R. Douglas, C.L. Yauk and F. Marchetti (2015), "Sublinear response in lacZ mutant frequency of Muta™ Mouse spermatogonial stem cells after low dose subchronic exposure to N-ethyl-N-nitrosourea", Environ. Mol. Mutagen., 56(4): 347-55.
Pegg, A.E. (2011), "Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools", Chem. Res. Toxicol., 24(5): 618-639.
Scherer, E., A.A. Jenner and L. den Engelse (1987), "Immunocytochemical studies on the formation and repair of O6-alkylguanine in rat tissues", IARC Sci. Publ., 84: 55-8.
Sega, G.A., C.R. Rohrer, H.R. Harvey and A.E. Jetton (1986), "Chemical dosimetry of ethyl nitrosourea in the mouse testis", Mutat. Res., 159(1-2): 65-74.
Seiler, F., K. Kamino, M. Emura, U. Mohr and J. Thomale (1997), "Formation and persistence of the miscoding DNA alkylation product O6-ethylguanine in male germ cells of the hamster", Mutat. Res., 385(3): 205-211.
Shrivastav, N., D. Li and J.M. Essignmann (2010), "Chemical biology of mutagenesis and DNA repair: cellular response to DNA alkylation", Carcinogenesis, 31(1): 59-70.
van Zeeland, A.A., A. de Groot and A. Neuhäuser-Klaus (1990), "DNA adduct formation in mouse testis by ethylating agents: a comparison with germ-cell mutagenesis", Mutat. Res., 231(1):55-62.
Wyatt, M.D., J.M. Allan, A.Y. Lau, T.E. Ellenberger, L.D. Samson (1999), "3-methyladenine DNA glycosylases: structure, function, and biological importance", Bioessays, 21(8): 668-676.
York S.J. and P. Modrich (2006), "Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts", J. Biol. Chem., 281(32): 22674-22683.
Relationship: 1910: Inadequate DNA repair leads to Increase, DNA strand breaks
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Oxidative DNA damage leading to chromosomal aberrations and mutations | adjacent | High | Low |
| Alkylation of DNA leading to decreased sperm count | adjacent | High | Moderate |
| Excessive reactive oxygen species leading to growth inhibition via oxidative DNA damage | adjacent | ||
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell cycle disruption | adjacent | High | Low |
| Reactive oxygen species leading to growth inhibition via oxidative DNA damage and cell death | adjacent | High | Moderate |
| Bulky DNA adducts leading to chromosomal aberrations and mutations | adjacent |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages |
| Sex | Evidence |
|---|---|
| Unspecific |
This KER applies to any cell type that has DNA repair capabilities.
Key Event Relationship Description
Inadequate repair of DNA damage includes incorrect repair (i.e., incorrect base insertion), incomplete repair (i.e., accumulation of repair intermediates such as strand breaks, stalled replications forks, and/or abasic sites), and absent repair resulting in the retention of DNA damage.
It is well-established that DNA excision repair pathways require DNA strand breakage for removing the damaged sites; for example, base excision repair (BER) of oxidative lesions involves removal of oxidized bases by glycosylases followed by cleavage of the DNA strand 5’ from the abasic site. If the repair process is disrupted at this point, repair intermediates including single strand breaks (SSB) may persist in the DNA. A SSB can turn into a double strand break (DSB) if it occurs sufficiently close to another SSB on the opposite strand. SSBs can be converted into DSBs when helicase unwinds the DNA strands during replication. Furthermore, SSBs and abasic sites can act as replication blocks causing the replication fork to stall and collapse, giving rise to DSBs (Minko et al., 2016; Whitaker et al., 2017).
The two most common DSB repair mechanisms are non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ is may favoured over HR and has also been shown to be 104 times more efficient than HR in repairing DSBs (Godwin et al., 1994; Benjamin and Little, 1992). There are two subtypes of NHEJ: canonical NHEJ (C‐NHEJ) or alternative non-homologous end joining (alt-NHEJ). During C-NHEJ, broken ends of DNA are simply ligated together. In alt‐NHEJ, one strand of the DNA on either side of the break is resected to repair the lesion (Betermeir et al., 2014). Although both repair mechanisms are error‐prone (Thurtle‐Schmidt and Lo, 2018), alt-NHEJ is considered more error-prone than C-NHEJ (Guirouil-Barbat et al., 2007; Simsek and Jasin, 2010). While NHEJ may prevent cell death due to the cytotoxicity of DSBs, it may lead to mutations and genomic instability downstream.
Evidence Supporting this KER
Biological Plausibility1. DNA strand breaks generated due to faulty attempted repair
Excision repair pathways require the induction of SSB as part of damage processing. Increases in DNA lesions may lead to the accumulation of intermediate SSB. Attempted excision repair of lesions on opposite strands can turn into DSBs if the two are in close proximity (Eccles et al., 2010). Generation of DSBs has been observed in both BER and nucleotide excision repair (NER) (Ma et al., 2009; Wakasugi et al., 2014).
Previous studies have demonstrated that an imbalance in one of the multiple steps of BER can lead to an accumulation of repair intermediates and failed repair. It is highly likely that a disproportionate increase in oxidative DNA lesions compared to the level of available BER glycosylases leads to an imbalance between lesions and the initiating step of BER (Brenerman et al., 2014). Accumulation of oxidative lesions, abasic sites, and SSBs generated from OGG1, NTH1, and APE1 activities would be observed as a result. Moreover, studies have reported accumulation of SSB due to OGG1- and NHT1-overexpression (Yang et al., 2004; Yoshikawa et al., 2015; Wang et al., 2018). BER repair intermediates have been observed to interfere with transcription as well (Kitsera et al., 2011). While overexpression may lead to imbalanced lyase activities that generate excessive SSB intermediates, deficiency of these enzymes is also known to cause an accumulation of oxidative lesions that could lead to strand breaks downstream. Hence, both the overexpression and deficiencies of repair enzymes can lead to strand breaks due to excessive activity or inadequate repair, respectively.
Similar to BER, NER involves coordinated dual incision of the DNA strand flanking bulky lesions, generating single-stranded DNA gaps as repair intermediates. If these intermediates persist due to incomplete repair or imbalance in downstream processing steps, they can contribute to the accumulation of strand breaks. For example, NER processing of UV-induced DNA damage can lead to the formation of DSBs, particularly through the persistence of single-stranded DNA gaps or secondary processing events (Wakasugi et al., 2014). In addition, structure-specific endonucleases involved in NER, such as XPF-ERCC1 and XPG, can introduce strand incisions that, when improperly coordinated or occurring at closely spaced lesions, may result in DSB formation (Riedl et al., 2003). Thus, dysregulation or saturation of NER can contribute to DNA strand break formation through the accumulation and improper resolution of repair intermediates.
2. DNA strand breaks generated due to replication stress caused by accumulated DNA lesions
Retention of DNA lesions (i.e., damaged bases and SSB) can interfere with the progression of the replication fork. Thymidine glycol is an example of an oxidative DNA lesion that acts as a replication block (Dolinnaya et al., 2013). Persistent replication fork stalling and/or dissociation of replication machinery are known to cause the replication fork to collapse, which generates highly toxic DSBs (Zeman and Cimprich, 2014; Alexander and Orr-Weaver, 2016). Fork stalling also increases the risk of two replication forks colliding with each other, generating DSBs.
Finally, the replication fork can collide with SSBs generated during BER, hindering the completion of repair and giving rise to DSBs (Ensminger et al., 2014).
DNA alkylation is one of the lesions that causes DNA strand breaks. O6-alkylguanine lesions are primarily repaired by O6-alkylguanine-DNA alkyltransferase (AGT, also known as O6-methylguanine-DNA methyltransferase MGMT in mammals), which is an established suicide protein that is irreversibly inactivated following a single repair event (Pegg, 2011; Fang et al, 2024). AGT/MGMT activity can become depleted, saturated, and insufficient to remove these lesions, particularly when exposed to high doses of alkylating agents. Consequently, O6-alkylguanine can persist in DNA and mispair with thymine during replication (Fahrer and Christmann, 2023; Fang et al, 2024). Subsequent mismatch repair (MMR) processing of O6-alkyl G:T mispairs generates repeated cycles of futile repair, leading to replication fork stalling, replication stress, and ultimately the formation of DNA DSBs (Roos and Kaina, 2006; Kaina et al., 2007; Fahrer and Christmann, 2023). Recent evidence suggests that interactions between rapid intermediates generated by BER, MMR, and replication-associated processing of alkylated DNA can also result in DSBs, even in non-dividing cells (Fujii and Fuchs, 2024). Collectively, these mechanism provides a well-characterized biological link between inadequate repair of alkylation-induced DNA lesions and the formation of DNA strand breaks.
In vitro studies with empirical evidence are shown below for select DNA repair pathways. These studies build in elements of essentiality (modulation of DNA repair), as well as dose and incidence concordance. The primary evidence is essentiality, where repair is genetically modulated in some way. Because multiple lines of evidence are considered within individual studies, we present the data by source of evidence (in vitro versus in vivo) rather than by type of empirical evidence (dose, incidence, or temporal concordance; essentiality) to avoid repetitive use of the same studies. This is a very data-rich area, and the examples presented here are intended to be illustrative rather than exhaustive, reflecting a broader and well-established body of evidence in which mechanistic understanding and essentiality provide strong overall support for this KER.
Inadequate repair of oxidative lesions
- Concentration concordance of strand breaks in repair-deficient and –proficient cells (insufficient repair) (Wu et al., 2008)
- In a study using A549 human adenocarcinoma cells, DNA strand breaks in hOGG1-proficient and hOGG1-deficient cells were compared following exposure to increasing concentrations of bleomycin.
- Strand breaks were measured as DNA migration length in alkaline comet assay after 3 hours of exposure to six increasing concentrations (0.05, 0.25, 0.5, 1, 5, and 10 mg/L).
- Concentration-dependent increase in strand breaks was observed in both cell types; however, at all concentrations significantly more strand breaks (p<0.05) were present in the hOGG1-deficient cells than in the proficient cells, demonstrating insufficient repair of oxidative lesions leading to DNA strand breaks.
- Thus, this evidence supports the essentiality of inadequate DNA repair as a modulator of the downstream KE.
- Incomplete OGG1-initiated base excision repair (BER) leads to DNA strand breaks (Wang et al., 2018):
- In a study using mouse embryonic fibroblasts (MEF), Ogg1+/+ and Ogg1-/- cells were treated with increasing concentrations of H2O2 for varying durations
Higher levels of 8-oxodG were detected in Ogg1-/- cells compared to Ogg1+/+ cells after treatment with 400 µM H2O2 at all time points (5, 15, 30, 60, and 90 min)- Demonstrates insufficient removal of 8-oxo-dG in OGG1-deficient cells
- Significantly more strand breaks, as indicated by the higher % of TUNEL-positive cells (p<0.001), were detected in Ogg1+/+ cells compared to Ogg1-/- cells after exposure to 400 µM H2O2 for 3 hours
- Both cell types showed a very similar increase in DNA strand breaks at lower concentrations (50, 100, and 200
µM) and there was no significant difference between Ogg1+/+ and Ogg1-/- cells at these concentrations – this suggests that up to a certain level of oxidative damage, OGG1-initiated BER does not exacerbate strand breaks but when oxidative stress is excessive (at 400µM in this study), OGG1-initiated BER is compromised and leads to increased strand breaks (incomplete repair)
- Both cell types showed a very similar increase in DNA strand breaks at lower concentrations (50, 100, and 200
- Finally, DNA strand breaks in both cell types were measured using both alkaline and neutral comet assay after a 30- minute exposure to 400µM H2O2; while there was an increase in the olive tail moment (indicating DNA strand breaks) in both cell types compared to the control, the increase of strand breaks in Ogg1+/+ cells was significantly larger than in Ogg1-/- cells in both assays (p<0.001)
- In a study using mouse embryonic fibroblasts (MEF), Ogg1+/+ and Ogg1-/- cells were treated with increasing concentrations of H2O2 for varying durations
Inadequate repair of alkylated DNA
- Interference of N-methylpurine DNA glycosylase (MPG)-initiated BER by replication leading to strand breaks (Ensminger et al., 2014)
- A549 human alveolar basal epithelial cells were exposed to increasing concentrations of methylmethane sulfonate (MMS) for 1 hour and replicating cells were labeled using a thymidine analogue, 5-ethynyl-2’-desoxyuridine (EdU).
- In S-phase cells, MMS concentration-dependent increase in γH2AX foci was detected (70 foci/cell at the highest concentration). In contrast, γH2AX foci were not detected G1- and G2-phase cells until the highest concentration (15 foci/cell).
- MPG-depleted cells in S-phase showed no significant increase in γH2AX foci, while the control cells showed significant MMS concentration-dependent increases.
- These results suggest interference of MPG-initiated BER by replication, leading to DSBs, and that the depletion of MPG decreases the probability of strand breaks in S-phase (evidence of essentiality of ‘inadequate repair’ to KEdown).
- Depletion of O6-alkylguanine-DNA alkyltransferase (AGT/MGMT) enhances the genotoxic effects of alkylating agents
- Numerous studies have demonstrated that genetic depletion or pharmacological inhibition of AGT/MGMT increases cellular sensitivity to alkylating chemotherapeutic agents (e.g., enhanced cytotoxicity, mutagenicity, and chromosomal damage) (Dolan et al., 1990, 1991; reviewed by Rabik et al., 2006). Although many of these studies did not directly measure DNA strand breaks, they provide strong evidence that AGT/MGMT-mediated repair is essential for preventing the persistence and biological consequences of O6-alkylguanine lesions.
- MGMT depletion increases alkylation-induced DNA strand breaks (direct evidence of essentiality)
- Roos et al. (2004) exposed human peripheral lymphocytes to methylating agents, N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) or temozolomide, following pharmacological inhibition of MGMT using O6-benzylguanine. In proliferating lymphocytes, MGMT depletion significantly increased DNA DSBs, as indicated by the neutral comet assay and γH2AX phosphorylation. Similar effects were induced by temozolomide. These findings provide direct evidence linking impaired MGMT-mediated repair to increased DNA strand breaks.
- Carlsson et al. (2025) exposed human HepG2-CYP2E1 cells to N-nitrosodimethylamine (NDMA), a methylating agent that generates O6-methylguanine after metabolic activation. NDMA induced a concentration-dependent increase in O6-methylguanine adducts 2 h post-exposure, with adduct levels persisting and further increasing at 24 h. NDMA also induced strand breaks, as indicated by the alkaline comet assay and increased γH2AX foci formation, and micronuclues formation. Inhibition of MGMT by O6-benzylguanine significantly enhanced these responses compared to NDMA treatment alone. In contrast, MGMT inhibition did not significantly affect DNA strand break formation induced by N-nitrosodiethanolamine (NDELA) or N-nitrosomethylaniline (NMA), consistent with differences in the types of DNA lesions generated by these compounds and indicating that the contribution of MGMT-mediated repair depends on lesion chemistry.
- These findings provide direct evidence that inadequate MGMT-mediated repair of O6-methylguanine lesions promotes the accumulation of downstream DNA strand breaks and chromosomal damage.
Inadequate mismatch repair
- Incomplete/incorrect mismatch repair (MMR) leads to DNA strand breaks (Peterson-Roth et al., 2005):
- MLH1 (MMR protein)-deficient and -proficient HCT116 human colon cancer cells were treated with 30µM K2CrO4 (DNA crosslinking, Cr adducts, protein-DNA crosslinking, DNA oxidation) for 3, 6, and 12 hours and γH2AX foci (biomarker of DNA DSB) were scored by fluorescence microscopy
- At 6 and 12 hours, MLH1+ cells had higher percentage of γH2AX foci than MLH1- cells
- The futile repair model of MMR suggests that strand breaks arise from MMR attempting repeatedly to repair the newly synthesized strand opposite adducts in S and G2 phases; approximately 80% of the γH2AX-positive MLH1+ cells were in G2 phase 12 hours after a 3-hour exposure to 20 µM Cr(VI), while the level was five times lower in MLH1- cells, suggesting that the MMR-induced DSB occurred following DNA synthesis; this supports the futile repair model and demonstrates inadequate repair
Inadequate Repair of DSBs
- Rydberg et al. [2005] exposed GM38 primary human dermal fibroblasts to increasing doses of linear electron transfer (LET) radiation of helium and iron ions (Rydberg et al., 2005).
- The cells were allowed to recover for 16 hours following irradiation.
- Unrepaired DSBs were measured after recovery using PFGE.
- There was a dose-dependent increase in unrepaired DSBs due to both ion exposures.
- Increase in persistent unrepaired DSBs with increasing dosage indicates exceeded repair capacity.
- DSB repair was also monitored by measuring γH2AX foci 0.05 - 24 hours after irradiation.
- DSBs decreased over time and less than 1 foci per cell on average remained in MRC-5 cells 24hours after 0.02, 0.2 and 2 Gy exposures.
- Repair was slower in 180BR cells, particularly for the 2 Gy exposure, where 20 foci per cell remained after 24 h.
- A follow-up study by the same group, found similar results for MRC-5 and 180BR cells exposed to 0.02 and 0.2 Gy of X-rays (Kühne et al., 2004).
- Rothkamm and Löbrich (2003) exposed MRC-5 primary human lung fibroblasts (repair-proficient) and 180BR DNA ligase IV-deficient human fibroblasts to 10 and 80 Gy of X-rays (Rothkamm and Lobrich, 2003).
- DNA ligase IV deficiency results in impaired NHEJ
- DSB repair was monitored using PFGE by measuring the % of DSBs remaining after 0.25, 2, and 24 h following irradiation.
- DSBs decreased over time and, eventually, less than 10% of the DSBs remained in MRC-5 cells after 24h following both 80 and 10 Gy exposures.
- Repair was noticeably slower in 180BR cells, where the clearance of DSBs was hindered and approximately 40 and 20% of the DSBs remained at 24 hours following 80 and 10 Gy exposures, respectively.
- The above demonstrates defective DNA repair leading to persistent DSBs.
Uncertainties and Inconsistencies
- A variety of confounding factors and genetic characteristics (i.e., SNPs) may modulate which repair pathways are invoked and the degree to which they are inadequate. These have yet to be fully defined.
- Both protective and damaging effects of OGG1 against strand breaks have been described in the literature. As demonstrated in the section above, the effect of OGG1-deficiency (BER-initiating enzyme) is observed to be different in different cell types; Wang et al. (2018) demonstrated strand breaks exacerbated by excessive OGG1 activity, while Wu et al. (2008) and Shah et al. (2018) demonstrated increased strand breaks due to lack of repair in mammalian cells in culture (Shah et al., 2018; Wu et al., 2008; Wang et al., 2018). Cell cycle and replication may influence the effect of DNA repair on exacerbating strand breaks.
- Dahle et al. (2008) exposed wild type and OGG1-overexpressing Chinese hamster ovary cells, AS52, to UVA. While OGG1-overexpression prevented the accumulation of Fpg-sensitive lesions (e.g., 8-oxo-dG and FaPyG) that were observed in wild type cells 4 hours after irradiation, there was no difference in the amount of strand breaks in the two cell types at 4h (Dahle et al., 2008).
- A recent study suggests that the NHEJ may be more accurate than previously thought (reviewed in Betermier et al., 2014). The accuracy of NHEJ may be dependent on the structure of the termini. The termini processing rather than the NHEJ itself is thus argued to be error-prone process (Betemier et al., 2014).
References
Alexander, J., Orr-Weaver, T. (2016), Replication fork instability and the consequences of fork collisions from rereplication, Genes Dev, 30:2241-2252.
Carlsson, M. J., Herzog, N., Felske, C., Ackermann, G., Regier, A., Wittmann, S., Cereijo, R. F., Sturla, S. J., Küpper, J.-H. & Fahrer, J. (2025). The DNA Repair Protein MGMT Protects against the Genotoxicity of N‑Nitrosodimethylamine, but Not N‑Nitrosodiethanolamine and N‑Nitrosomethylaniline, in Human HepG2 Liver Cells with CYP2E1 Expression. Chemical Research in Toxicology, 38(6), 1134–1146. https://doi.org/10.1021/acs.chemrestox.5c00133
Dolan, M. E., Moschel, R. C. & Pegg, A. E. (1990). Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proceedings of the National Academy of Sciences, 87(14), 5368–5372. https://doi.org/10.1073/pnas.87.14.5368
Dolan, M. E., Mitchell, R. B., Mummert, C., Moschel, R. C. & Pegg, A. E. (1991). Effect of O6-benzylguanine analogues on sensitivity of human tumor cells to the cytotoxic effects of alkylating agents. Cancer Research, 51(13), 3367–3372.
Brenerman, B., Illuzzi, J., Wilson III, D. (2014), Base excision repair capacity in informing healthspan, Carcinogenesis, 35:2643-2652.
Dahle, J., Brunborg, G., Svendsrud, D., Stokke, T., Kvam, E. (2008), Overexpression of human OGG1 in mammalian cells decreases ultraviolet A induced mutagenesis, Cancer Lett, 267:18-25.
Dolinnaya, N., Kubareva, E., Romanova, E., Trikin, R., Oretskaya, T. (2013), Thymidine glycol: the effect on DNA molecular structure and enzymatic processing, Biochimie, 95:134-147.
Eccles, L., Lomax, M., O’Neill, P. (2010), Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage, Nucleic Acids Res, 38:1123-1134.
Ensminger, M., Iloff, L., Ebel, C., Nikolova, T., Kaina, B., Lobrich, M. (2014), DNA breaks and chromosomal aberrations arise when replication meets base excision repair, J Cell Biol, 206:29.
Kaina, B., Christmann, M., Naumann, S. & Roos, W. P. (2007). MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair, 6(8), 1079–1099. https://doi.org/10.1016/j.dnarep.2007.03.008
Kitsera, N., Stathis, D., Luhnsdorf, B., Muller, H., Carell, T., Epe, B., Khobta, A. (2011), 8-Oxo-7,8-dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by OGG1, Nucleic Acids Res, 38:5926-5934.
Kühne, M., E. Riballo, N. Rief, K. Rothkamm, P. Jeggo, & M. Löbrich (2004), "A Double-Strand Break Repair Defect in ATM-Deficient Cells Contributes to Radiosensitivity", Cancer Res, 64(2): 500-508.
Ma, W., Panduri, V., Sterling, J., Van Houten, B., Gordenin, D., Resnick, M. (2009), The Transition of Closely Opposed Lesions to Double-Strand Breaks during Long-Patch Base Excision Repair Is Prevented by the Coordinated Action of DNA Polymerase and Rad27/Fen1 , Mol Cell Biol, 29:1212-1221.
Minko, I., Jacobs, A., de Leon, A., Gruppi, F., Donley, N., Harris, T., Rizzo, C., McCullough, A., Lloyd, R.S. (2016), Catalysts of DNA Strand Cleavage at Apurinic/Apyrimidinic Sites, Sci Rep, 6.
Peterson-Roth, E., Reynolds, M., Quievryn, G., Zhitkovich, A. (2005), Mismatch Repair Proteins Are Activators of Toxic Responses to Chromium-DNA Damage, Mol Cell Biol, 25:3596-3607.
Pegg, A. E. (2011). Multifaceted Roles of Alkyltransferase and Related Proteins in DNA Repair, DNA Damage, Resistance to Chemotherapy, and Research Tools. Chemical Research in Toxicology, 24(5), 618–639. https://doi.org/10.1021/tx200031q
Rabik, C. A., Njoku, M. C. & Dolan, M. E. (2006). Inactivation of O6-alkylguanine DNA alkyltransferase as a means to enhance chemotherapy. Cancer Treatment Reviews, 32(4), 261–276. https://doi.org/10.1016/j.ctrv.2006.03.004
Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J.2003 Oct 1;22(19):5293-303. doi: 10.1093/emboj/cdg489.
Roos, W., Baumgartner, M. & Kaina, B. (2004). Apoptosis triggered by DNA damage O6-methylguanine in human lymphocytes requires DNA replication and is mediated by p53 and Fas/CD95/Apo-1. Oncogene, 23(2), 359–367. https://doi.org/10.1038/sj.onc.1207080
Roos, W. P. & Kaina, B. (2006). DNA damage-induced cell death by apoptosis. Trends in Molecular Medicine, 12(9), 440–450. https://doi.org/10.1016/j.molmed.2006.07.007
Rothkamm, K., Lobrich, M. (2003), Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses, Proc Natl Acad Sci USA, 100:5057-5062.
Rydberg, B., Cooper, B., Cooper, P., Holley, W., Chatterjee, A. (2005), Dose-Dependent Misrejoining of Radiation-Induced DNA Double-Strand Breaks in Human Fibroblasts: Experimental and Theoretical Study for High- and Low-LET Radiation, Radiat Res, 163:526-534.
Shah, A., Gray, K., Figg, N., Finigan, A., Starks, L., Bennett, M. (2018), . Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis, Circulation, 138:1446-1462.
Wakasugi, M., Sasaki, T., Matsumoto, M., Nagaoka, M., Inoue, K., Inobe, M., Horibata, K., Tanaka, K., Matsunaga, T. (2014), Nucleotide Excision Repair-dependent DNA Double-strand Break Formation and ATM Signaling Activation in Mammalian Quiescent Cells, J Biol Chem, 289:28730-28737.
Wang, R., Li, C., Qiao, P., Xue, Y., Zheng, X., Chen, H., Zeng, X., Liu, W., Boldogh, I., Ba, X. (2018), OGG1-initiated base excision repair exacerbates oxidative stress-induced parthanatos, Cell Death and Disease, 9:628.
Whitaker, A., Schaich, M., Smith, M.S., Flynn, T., Freudenthal, B. (2017), Base excision repair of oxidative DNA damage: from mechanism to disease, Front Biosci, 22:1493-1522.
Wu, M., Zhang, Z., Che, W. (2008), Suppression of a DNA base excision repair gene, hOGG1, increases bleomycin sensitivity of human lung cancer cell line, Toxicol App Pharmacol, 228:395-402.
Yang, N., Galick, H., Wallace, S. (2004), Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks, DNA Repair, 3:1323-1334.
Yoshikawa, Y., Yamasaki, A., Takatori., K., Suzuki, M., Kobayashi, J., Takao, M., Zhang-Akiyama, Q. (2015), Excess processing of oxidative damaged bases causes hypersensitivity to oxidative stress and low dose rate irradiation, Free Radic Res, 49:1239-1248.
Zeman, M., Cimprich, K. (2014), Causes and Consequences of Replication Stress, Nat Cell Biol, 12:2-9.
Relationship: 3801: Increase, DNA strand breaks leads to Apoptosis
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Alkylation of DNA leading to decreased sperm count | adjacent | High | Moderate |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages | High |
| Sex | Evidence |
|---|---|
| Unspecific | High |
This KER applies to eukaryotic cells exposed to genotoxic stressors that induce persistent or unrepaired DNA strand breaks. However, the applicability of this KER is context-dependent and influenced by cell type, developmental stage, DNA repair capacity, and DDR signaling status.
Key Event Relationship Description
DNA strand breaks activate a series of DNA damage response (DDR) pathways that together determine whether damaged cells go through cell cycle arrest, repair, or cell death. When the levels of DNA damage exceed the DNA repair capacity, DDR signaling shifts the cellular response toward elimination of damaged cells by various forms of cell death. Thus, DNA strand breaks are potent inducers of programmed cell death (i.e., apoptosis) when damage is irreparable or sustained.
Evidence Supporting this KER
The empirical support for this KER is strong based on a large body of literature connecting DNA strand breaks and the induction of apoptosis. Both SSBs and DSBs are frequently associated with increased apoptosis in a dose-concordant manner in somatic and germ cells, as well as animal models (Roos and Kaina, 2006). This concordance has been observed following exposures to many DNA-damaging agents; select examples in different tissues and models are provided below.
Evidence in vivo (exposure)
Saha et al. (2014) demonstrated clear temporal and dose concordance between DSB formation and apoptosis in the embryonic mouse brain. Mice were exposed in utero to acute doses of 10, 25, 50, 100 or 200 mGy X-rays, and embryo brains were examined 1 hour or 6 hour later. The number of 53BP1 foci per cell was quantified as a marker of DSB induction, while TUNEL staining (Terminal deoxynucleotidyl UTP Nick End Labeling) was used to monitor apoptosis. Significant increases in 53BP1 foci formation (at 1 hour) and TUNEL+ apoptotic cells (at 6 hours) were observed starting at 10 mGy, indicating that the two KEs occur within the same dose range and that DSBs occur earlier than apoptosis. At 6 hours post-irradiation, while the number of 53BP1 foci at lower doses remained relatively unchanged, it was reduced at50 mGy and returned to the control level at 100 mGy, indicating efficient repair. Despite this apparent resolution of DNA damage, apoptosis continued to increase in a dose-dependent manner, suggesting that even low and transient DSBs are sufficient to induce apoptosis in this sensitive tissue (Saha et al., 2014).
Acetaminophen (200 mg/kg) induces DNA fragmentation and apoptosis in hepatocytes after intraperitoneal administration in mice (Bajt et al., 2011). Liver cells collected from exposed mice showed the release of histone-associated DNA fragments, mitochondrial pro-apoptotic proteins (apoptosis-inducing factor AIF, Smac, endonuclease G, and cytochrome c) into the cytosol at 6 hours post-treatment, accompanied by increased TUNEL+ staining at both 6 and 24 hours. This study also demonstrates the essentiality of mitochondrial pathways through the use of AIF-deficient mice. When treated with acetaminophen, these mice showed a marked reduction in presence of DNA strand breaks (TUNEL assay), % DNA fragmentation, and the expression of apoptotic markers compared to wild-type counterparts. This indicates that AIF is a necessary mediator of DNA fragmentation and apoptosis (Bajt et al., 2011). However, detection of DNA fragments does not allow discrimination between upstream DNA damage and downstream apoptotic DNA fragmentation (see Uncertainties and Inconsistencies). As only a single dose and timepoint were assessed for most markers, the data primarily support co-occurrence.
Evidence in vivo (genetic manipulation)
Using Nestin-Cre mice, Rodrigues et al. (2013) showed that deletion of Nibrin (Nbn, a key sensor of DSBs) and Atm kinase in neural stem/progenitor cells led to DSB accumulation (γH2AX+ cells, p53 stabilization) and concordantly enhanced apoptosis (TUNEL+ cells and cleaved caspase-3 staining) in specific regions of the developing brain (cerebellum, ganglionic eminences) and eye (lens) on embryonic days E15.5 and E17.5. The incidence of γH2AX+ cells (~6,000 to 10,000 cells/mm2) was consistently higher than that of TUNEL+ cells (~250 to 800 cells/mm2) and caspase-3+ cells (~200 to 800 cells/mm2) in the embryonic brains (Figure 1). A similar pattern was observed in eye lens and retina, supporting incidence concordance. Greater impairment of DDR signaling (Nbn/AtmNes-Cre double deletion) induced more DSBs and higher apoptosis than Nbn single deletion alone (NbnNes-Cre) (Figure 1). This establishes a graded response-response relationship between DSB formation and apoptosis (Rodrigues et al., 2013). However, in the retina region, apoptosis induced by Nbn deletion was prevented by simultaneous inactivation of ATM, despite the presence of DNA damage. These findings provide direct evidence of essentiality, showing that key signaling pathways downstream of DNA strand breaks are required for the induction of subsequent apoptotic cell death. The observed differences across tissue suggest that these responses are context-dependent and may be mediated by tissue-specific pathways (Rodrigues et al., 2013).
Loss of Apc (adenomatous polyposis coli) in mouse liver tissue led to an early increase in DSBs, as indicated by elevated γH2AX and Rad51 staining at day 4, accompanied by upregulation of DNA damage checkpoint proteins p53 and p21 (Méniel et al., 2015). This was followed by a subsequent increase in apoptotic cells at day 6. Quantitative immunohistochemistry analysis revealed that the incidence of γH2AX-positive and Rad51-positive cells was consistently higher than that of caspase-3-positive apoptotic cells. At day 4, γH2AX-positive cells increased from 0.9% in WT to 26.2% in Apc-deficient mice and 67.1% in Apc/p53 double knockout mice, while Rad51 increased from 2% in WT to 9.9% and 20.7% after genetic manipulation (Figure 2). At the same timepoint, caspase-3-positive cells remained low (~0.36-1.34% across genotypes). A similar pattern was observed at day 6. Additional deletion of p53 further increased markers of DNA damage, indicating that p53 plays an essential role in regulating DSB levels following Apc loss. Together, these findings support incidence concordance and temporal sequence in which DSBs precede activation of checkpoint signaling and subsequent induction of p53-dependent apoptosis.
Xiao et al. (2023) established a myocardial ischemia/reperfusion mouse model to study the role of a long non-coding RNA (cardiac ischemia reperfusion associated Ku70 interacting lncRNA, or CIRKIL) in coronary artery disease. Overexpression of CIRKIL in mouse hearts and 100 μmol/L H2O2-treated mouse cardiomyocytes induced parallel increases in DSBs (γH2AX expression), p53 activation, and apoptotic markers, including the ratio of pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins (Bax/Bcl-2), caspase-3 activity, and TUNEL+ cells. Conversely, knockdown of CIRKIL, as well as single knockout or double knockout of CIRKIL and its partner Ku70, alleviated these myocardial injury and reduced markers of both DNA strand breaks and apoptosis. The consistent bidirectional changes in these endpoints by multiple genetic interventions supports the role of the CIRKIL/Ku70 pathway in linking DNA strand breaks to apoptotic responses, and provides indirect evidence of essentiality (Xiao et al., 2023).
Together, the in vivo evidence provides strong empirical support for temporal concordance between strand breaks and apoptosis. Concurrent changes in both KEs were often observed at the same dose level, supporting qualitative dose concordance. A few studies support incidence concordance. In addition, the data highlight that the progression from DNA strand breaks to apoptosis is influenced by tissue-specific sensitivity and mechanisms, which should be considered when evaluating the applicability of this KER.
Evidence in vitro
Roos et al. (2004) provided important mechanistic evidence linking DNA alkylation, impaired repair, DNA strand breaks, and apoptosis in human lymphocytes. O6-methylguanine (O6-MeG) is a key DNA lesion associated with apoptosis, and its removal is mediated by O6-MeG-DNA methyltransferase (MGMT). To prevent repair of O6-MeG, cells were pre-treated with O6-benzylguanine to inactivate MGMT, followed by treatment with O6-MeG-generating agents, N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) or an anticancer drug temozolomide. In MGMT-depleted proliferating lymphocytes, O6-MeG lesions were processed by mismatch repair during DNA replication, leading to the formation of DSBs, as demonstrated by neutral comet assay and γH2AX expression analysis. MNNG (10 or 25 μM) induced DSBs within 3 hours and the level peaked at 24 hours post-exposure. Apoptosis (measured by sub-G1 DNA content analysis or Annexin V/PI staining) was only observed after 24 hours (and persisted up to 96 hours) following MNNG treatment (at multiple concentrations, up to 20 μM), occurring later than DSB formation. Similar effects were induced by temozolomide. Apoptosis was associated with p53 stabilization and upregulation of the Fas receptor, and was significantly reduced (~61%) by treatment with an anti-Fas neutralizing antibody. In contrast, non-proliferating cells showed minimal apoptosis at lower MNNG concentrations (≤ 10 μM), indicating that DNA replication and mismatch repair is required in converting O6-MeG to DSBs. Furthermore, ionizing radiation (5 Gy), which directly induces DSBs, triggered apoptosis in both proliferating and non-proliferating lymphocytes. Together, these data suggest that O6-MeG is converted during DNA replication into DSBs that trigger apoptosis via p53- and Fas-dependent pathways. In addition, MGMT inactivation provides direct evidence of essentiality, as impaired DNA repair enhances the persistence of DNA strand breaks and contributes to apoptosis.
He et al. (2018) studied the effects of loperamide (an antidiarrheal agent) on DNA damage and apoptosis in leukemia cell lines and human primary leukemia cells. Cells exposed to 5, 10, 20 μM loperamide for 24 hours exhibited clear evidence for DNA strand breaks, which were detected by the alkaline comet assay (identifying SSBs and alkali-labile sites) and increased expression of γH2AX. In the same concentration range, a dose-dependent increase in the percentage of apoptotic cells was quantified by Annexin V/7-aminoactinomycin D+ staining and flow cytometry. The induction of apoptosis was further confirmed by increased protein expression of two apoptotic markers, cleaved-caspase-3 and cleaved poly (ADP‑ribose) polymerase (PARP), and decreased expression of an anti‑apoptosis protein Mcl‑1 at 10 μM and 20 μM. Activation of the ATM‑CHK2 pathway following 5‑20 µM loperamide treatment supported the induction of a DDR pathway in leukemia cells. As an example, the effects of loperamide on strand break formation and apoptosis in Molt-4 acute lymphocytic leukemia cells are shown in Figure 3. Similar effects were observed in Thp1 acute myelocytic leukemia cells and primary human leukemia cells (He et al., 2018).
Similarly, exposure to quercetin (40-80 μM) and the positive control paclitaxel (0.45 μM) for 48 hours induced simultaneous strand breaks and apoptosis in MGMT-positive human glioblastoma T98G cells (Wang et al., 2023). DNA strand breaks were assessed using the alkaline and neutral comet assays, and γH2AX staining, capturing both SSBs and DSBs. Apoptosis was assessed by the TUNEL assay, Annexin V-FITC and PI double staining, the ratio of Bax/Bcl-2, and the expression of apoptosis-related proteins, including cleaved caspase-9, cleaved caspase-3, and cleaved PARP. Quercetin (starting at 40 μM) and paclitaxel induced concentration-dependent increases in tail moment (tail length × % tail DNA) in the comet assays and γH2AX expression, indicating DSB formation. At the same concentrations and timepoint, both compounds significantly increased apoptotic cells, accompanied by a higher Bax/Bcl-2 ratio and elevated expression of apoptosis-related markers. Notably, quercetin (≥ 40 μM) also downregulated MGMT expression and activity, suggesting reduced MGMT-mediated DNA repair (Wang et al., 2023). These data support dose and temporalconcordance between DNA strand breaks and apoptosis; the concurrent downregulation of MGMT supports a role of insufficient DNA repair in the accumulation of DNA strand breaks and subsequent apoptosis.
Yauk et al. (2016) studied micronucleus induction and apoptosis in human lymphoblastoid TK6 cells following 4-hour exposures to six genotoxic chemicals: 2-aminoanthracene (0.75-2 μg/mL), acetaminophen (1-10 mM), cyclophosphamide (1-20 μM), dibenz[a,h]anthracene (1-280 μg/mL), dimethylnitrosamine (1-10 mM), furan (2-5 mM) (Figure 4). Micronucleus frequency serves as a marker of chromosomal damage resulting from unrepaired DNA lesions (including DSBs), but it is not a direct measure of DNA strand breaks. Consistent dose-dependent increases in both micronucleus frequency and the number of apoptotic cells were observed 20 hours post-exposure across all tested chemicals (Figure 4). In general, significant increases in apoptosis occurred at slightly lower concentrations than those required to induce micronucleus formation, and fewer cells exhibited chromosome damage than apoptosis, indicating a lack of incidence and only partial dose concordance. These observations likely reflect differences in assay sensitivity and biological progression, as micronucleus formation is a late-stage cytogenetic outcome that requires progression through mitosis. Therefore, these differences do not contradict the overall relationship. As both endpoints were measured at the same time, these data support temporal concordance of this KER.
Liu et al. (2014) showed that inhibition of the PI3K/AKT pathway (a regulator of DDR) by BKM120 led to DNA damage and increased apoptosis in human and murine hepatocellular carcinoma cell lines (Huh7 and BNL). Cells pretreated with 1 μM BKM120 for 1 hour were irradiated (10 Gy for Huh7 cells and 5 Gy for BNL cells). Radiation induced DSB formation (assessed by γH2AX staining) within 30 minutes and the presence of the PI3K inhibitor led to prolonged foci retention (≥ 8 hours in Huh7 cells and ≥ 4 hours in BNL cells). In parallel, PI3K inhibition increased radiation-induced apoptosis, as indicated by elevated cleaved caspase-3 expression and increased Annexin V-positive cells at 24 hours. Co-treatment with rapamycin, an mTOR inhibitor downstream of PI3K, further enhanced both DSB persistence and apoptosis (Liu et al., 2014). These findings demonstrate strong temporal concordance, with DSBs occurring earlier than apoptotic responses; parallel increases were observed across experimental conditions. Although dose-response data are not available, modulation of DNA damage persistence through PI3K/AKT and downstream mTOR inhibition provides indirect essentiality support for the link between DNA strand breaks and apoptosis.
Additional empirical evidence from Chen et al. (2022) supports this KER in human nasopharyngeal carcinoma cells. Two cell lines, SUNE1 and HONE1, were transfected to overexpress ubiquitin-specific protease 44 (USP44). USP44 regulates ubiquitin signaling at DSB sites, thereby modulating DDR and repair processes. Following irradiation (6 Gy), both cell lines exhibited rapid induction of DNA strand breaks, as shown by significant increases in tail moment in the neutral comet assay and higher γH2AX foci numbers within 30 minutes to 4 hours post-exposure. Minimal or no DNA damage was observed at 24 hours, indicating efficient repair at later timepoints. In the same system, USP44 overexpression promoted radiation-induced apoptosis, demonstrated by Annexin V/PI staining at 24 hours in vitro and increased caspase-3 staining in subcutaneous xenograft tumors in mice (Chen et al., 2022). The alignment between early DSB formation and subsequent apoptotic responses supports strong temporal concordance; concurrent increases in both KEs following USP44 overexpression provide indirect essentiality evidence for this KER.
CDKN2AIP is a cell cycle-associated protein that has been implicated in the regulation of DNA damage repair, p53-dependent apoptosis, and spermatogenesis. An in vitro study by Cao et al. (2022) using Cdkn2aip-knockdown TM4 mouse testicular Sertoli cells exposed to γ-irradiation (5 Gy) showed impaired DSB repair, evidenced by persistent γH2AX and 53BP1 foci formation at 2h, 12h, and 24h, together with a significant increase in apoptosis measured by flow cytometry and TUNEL at 24h (Cao et al., 2022). This study provides evidence for temporal concordance and essentiality.
Overall, across multiple somatic cell types, radiation-induced, chemically or genetically induced DNA strand breaks are consistently associated with an increase in apoptosis. These studies demonstrate strong temporal concordance, with DNA strand breaks occurring early or concurrently with apoptotic responses. Evidence for dose concordance is supported in several studies but dose-response data are not always available. Evidence for incidence concordance is limited in vitro. The consistent direction of responses across experimental conditions provide strong empirical support for the relationship between these two KEs in somatic cells.
Evidence in germ cells
A study by Hamer et al., (2003) provided evidence linking DSBs and apoptotic responses in male germ cells. They first demonstrated the presence of basal γH2AX in testes of wild-type FvB/NAU mice. Following a single ionizing radiation (4 Gy), γH2AX foci were rapidly (within 3 hours) induced in spermatogonia, spermatocytes, and round spermatids. In spermatogonia, γH2AX and p53 colocalized in the nucleus and their interaction was enhanced following irradiation. These findings support co-occurrence of DSBs and p53-dependent apoptosis in spermatogonia.
Habas et al. reported the effects of known mutagens on testicular germ cells isolated from 10-12-week-old NMRI mice (National Medical Research Institute) or male Sprague-Dawley rats. Isolated testicular germ cells (spermatogonia, spermatocytes and spermatids) were exposed for 1 hour to one of seven genotoxic compounds at 50, 500, or 1000 μM, and were immediately subjected to alkaline comet or TUNEL analyses. The compounds included doxorubicin (Dox), which induces DNA strand breaks via topoisomerase II inhibition (Habas et al., 2017), and a panel of mutagens targeting germ cells at specific stages: alkylating agents N-ethyl-N-nitrosourea (ENU) and N-ethyl-N-nitrosourea (MNU), which target pre-meiotic spermatogonia; 6-mercaptopurine (6-MP) and 5-bromo-20-deoxy-uridine (5-BrdU), which produce the greatest responses in early meiotic spermatocytes; and methyl methanesulphonate (MMS) and ethyl methanesulphonate (EMS) that primarily target post-meiotic germ cells (Habas et al., 2016). Representative results (Figure 5) show highly consistent dose-dependent increases in both % tail DNA in the alkaline comet assay and the percentage of TUNEL+ apoptotic cells across male germ cells at all stages, with both effects being statistically significant at the same exposure concentrations (Habas et al., 2016, 2017). These data support dose and temporal concordance while extending the applicability of this KER to male germ cells.
Cao et al. (2022) demonstrated that loss of CDKN2AIP leads to a concurrent increase in DNA strand breaks and apoptosis in male germ cells. CDKN2AIP preferentially expresses in spermatocyte and spermatids and acts as a central regulator of DSBs repair. Analyses of Cdkn2aip-/- male C57BL/6 mice revealed a significant accumulation of DSBs (γH2AX analyses) in pachytene spermatocytes from postnatal day P35 testes, along with a higher incidence of TUNEL foci per cells in the seminiferous tubules (Cao et al., 2022). These data indicate a clear association between increased DSBs and male germ cell apoptosis, and provide evidence for the essential role of DNA damage repair in preventing DSB accumulation and subsequent apoptotic cell death.
Suh et al. (2006) examined oocytes from Balb/c mice exposed to γ-irradiation (0, 0.1, 0.45 Gy) on postnatal day 5. At 24 hours post-exposure, on average, oocytes exposed to 0.1 Gy had an average of three γH2AX-marked DSBs with minimal apoptosis; oocytes receiving 0.45 Gy had about ten DSBs and underwent apoptosis within 24 hours, as indicated by condensed chromatin and positive TUNEL staining (Suh et al., 2006). Consistent with early DNA damage induction, Stringer et al. (2020) reported dose- and time-dependent increases in nuclear localization of phosphorylated ATM in mouse oocytes as early as 30 minutes following irradiation (0.2, 0.45, 7 Gy), indicating activation of DNA damage responses. The presence of DSBs was evidenced by the formation of γH2AX foci at all doses. Apoptosis was indirectly monitored by follicle survival at 24 hours and oocyte retrieval rate. At the same timepoints and dose range, Rad51 (a marker of homologous recombination) localized to DNA damage sites in over 90% of oocytes in primordial follicles, whereas DNA-dependent protein kinase catalytic subunit (DNA-PKcs, a marker of non-homologous end joining) was mostly present in granulosa cells; this difference suggests cell type-specific utilization of repair pathways. Importantly, intervention studies in this system provide indirect evidence of essentiality. In Tap63 knockout mice (deficient in DNA damage-induced apoptotic signaling), primordial follicles were preserved despite the presence of DNA damage. Conversely, pharmacological inhibition of Rad51 increased persistence of unrepaired DNA damage and apoptosis, suggesting that efficient DNA repair mitigated the progression from DNA strand breaks to cell death.
Experimental evidence from isolated germ cells demonstrates that DNA strand breaks are closely associated with increased apoptosis across multiple stages of spermatogenesis and in oocytes. These findings support the applicability of this KER to germ cells and reinforce dose and temporal concordance.
Biological PlausibilityThe biological plausibility of this KER is strong and supported by extensive understanding of the DDR pathways (Jackson and Bartek, 2009). The mechanistic processes that link DNA strand breaks to apoptosis are well characterized and detailed in many excellent reviews including Kaina (2003), Roos and Kaina (2006), and Zio et al. (2012). Both p53-dependent (encoded by Tp53) and p53-independent mechanisms contribute to DNA strand break-induced apoptosis (Roos and Kaina, 2006). It is important to note that activation of p53 and other DDR pathways does not immediately trigger apoptosis. Rather, the cellular response depends on the extent and persistence of DNA damage. In many cases, initial DDR activation promotes cell cycle arrest and DNA repair (reviewed by Williams and Schumacher, 2016). When DNA damage is extensive, irreparable, or persistent, the DDR signaling cascade shifts to favour programmed cell death, in part through the action of p53 and other stress sensors (Maréchal and Zou, 2013). This conditional response reflects a protective mechanism that preserves genomic integrity and prevents the propagation of cells with damaged DNA.
Sensing of DNA strand breaks by serine/threonine kinases
The serine/threonine kinases, ATM (ataxia telangiectasia mutated) and ATR (ATM- and Rad3-Related), are key sensors and mediators of cellular responses to DNA strand breaks in cells (Maréchal and Zou, 2013).
ATM is predominantly activated by double strand breaks (DSBs). The MRN (Mre11- Rad50-Nbs1) complexrecognizes DSB ends and recruits ATM to the damage sites (Maréchal and Zou, 2013). In undamaged cells, ATM exists predominantly as inactive dimers/oligomers; in the presence of DSBs, ATM undergoes autophosphorylation anddissociates into active monomers that phosphorylate a series of proteins involved in cell cycle arrest, DNA repair, and apoptosis, such as CHK2, Brca1, H2AX, and p53 (Lee, 2005). Of these, phosphorylation of histone H2AX at serine 139 (γH2AX) is one of the earliest cellular responses to DSBs. γH2AX foci form rapidly (within minutes) at sites of damage and are widely recognized as a sensitive marker of DNA damage following chemical exposure in both somatic cells and germ cells (Hamer et al., 2003). In the testis, basal γH2AX signaling is observed across multiple stages of germ cells and is associated with p53-mediated apoptosis signaling in spermatogonia following stress (Hamer et al., 2003).
ATR kinase, on the other hand, responds to a broader spectrum of DNA damage. ATR is activated by replication protein A (RPA)-coated single-stranded DNA (ssDNA), which arises during replication stress or as intermediates in DNA repair (Maréchal and Zou, 2013). ATR activation requires its binding partner, ATRIP (ATR-interacting protein), which recognizes RPA-coated ssDNA and localizes ATR to the damage sites (Maréchal and Zou, 2013). Both single strand breaks (SSBs) and DSBs (when resected by nucleases) can generate ssDNA and thus activate ATR (Maréchal and Zou, 2013).
p53-dependent DNA strand break-triggered apoptosis
Both ATM and ATR kinases phosphorylate and activate p53 transcription factor, a master regulator of apoptosis.ATM primarily phosphorylates p53 at serine-15, whereas ATR targets both serine-15 and serine-37 (Tibbetts et al., 1999).These partially overlapping activities ensure p53 activation during genotoxic stress. Once p53 is activated by ATM/ATR, it promotes the transcription of genes that favour apoptosis over survival, particularly when DNA damage is persistent or irreparable. To accomplish this, p53 activates pro-apoptotic genes (e.g., Puma, Bax, Apaf-1, Noxa), while inactivating anti-apoptotic factors such as members in the Bcl-2 family. These pro-apoptotic factors increase the permeability of the mitochondrial membrane and signal the release of cytochrome c (Wawryk-Gawda et al., 1998). Cytochrome c then binds Apf-1 adaptor and procaspase-9, aggregating the proteins and activating caspase-9, which in turn activates caspase-3 to induce apoptosis (Elmore, 2007).
p53-independent DNA strand break-triggered apoptosis
Cells can also respond to DNA strand breaks and undergo apoptosis through several p53-independent mechanisms (Roos and Kaina, 2006). DNA strand breaks induce ATM/ATR-dependent phosphorylation of checkpoint proteins CHK1 or CHK2, which can trigger apoptosis independently of p53 by stimulating E2F1-dependent expression of pro-apoptotic factor p73 (Urist et al., 2004). A newly identified p53-independent apoptosis pathway is triggered by DNA strand breaks through ribosome stalling mediated by Schlafen 11 (SLFN11, a tRNase). Upon DNA damage, SLFN11 causes ribosome stalling and global translation inhibition. The stalled ribosomes are recognized by ZAKα, a ribosome-associated stress sensor, which then initiates a MAPK signaling cascade that directly activates the mitochondrial apoptosis machinery, independent of the canonical p53 pathway (Boon et al., 2024). Other factors may be indirectly activated by DNA strand breaks and contribute to p53-independent apoptosis, such as pathways involving Bcl-2 degradation, caspase-2 activation, and NF-κB dependent Fas ligand transcription (Roos and Kaina, 2006).
In the female germ line, p63, a homologue of p53, acts in a conserved mechanism to preserve genomic fidelity. Specifically, a p63 isoform TAp63 is essential in the process of DNA damage-induced oocyte death not involving p53 (Suh et al., 2006). TAp63 is constitutively expressed in oocytes during meiotic arrest. In response to DNA damage, TAp63 is phosphorylated and binds to p53 cognate DNA sites and results in transcriptional activation of apoptosis pathways in female germ line (Suh et al., 2006).
Uncertainties and InconsistenciesA primary uncertainty in this KER arises from the use of the TUNEL assay, which can detect both the primary DNA strand breaks and the secondary DNA fragmentation that occurs during late-stage apoptosis, necrosis, or severe oxidative stress. This overlap makes it difficult to distinguish whether the DNA damage is an initiating event or a downstream consequence, particularly when both KEs are measured at the same timepoint (Bajt et al., 2011). For stressors like acetaminophen that can induce extensive necrosis, interpreting the TUNEL signal as a specific marker of apoptosis may confound the causal interpretation of this KER. To reduce this uncertainty, we recommend studies using multiple timepoints to establish temporal concordance, and using direct DNA damage assays (e.g., comet assays, γH2AX staining) and apoptosis specific markers (e.g., caspase activation or Annexin V staining) to strengthen confidence in the conclusions.
This KER is context-specific. Factors like developmental stage, activity of the repair pathways, and cellular sensitivity can modulate this KER. During ovarian reserve formation, meiotic germ cells naturally accumulate high levels of programmed DSBs that are efficiently repaired. In female FVB/N mice, Zhou et al. (2021) reported increased levels of DSBs from embryonic day 13.5 to postnatal day 2, with 23.94% to 83.76% germ cells stained positive for γH2AX. Despite the high incidence of DSBs, no detectable germ cell apoptosis was observed based on caspase-3 and TUNEL staining. A similar phenomenon is observed in the male germ line. During spermatogenesis, programmed DSBs occur during meiosis and chromatin remodeling processes (e.g., histone-to-protamine replacement during spermiogenesis) (Talibova et al., 2022) . Together, these observations indicate that the apoptotic response is not determined by the presence of DSBs per se, but their persistence and repair failure are critical modifiers.
Moreover, some studies report apoptosis in the absence of detectable DNA strand breaks. For example, in mice exposed daily to furan for 28 days, no treatment-related induction of DNA strand breaks or DNA cross links was noted in liver cells, as measured by the alkaline comet assay and γH2AX foci formation at 24 hours after last administration (Cordelli et al., 2010). However, a significant increase in TUNEL+ apoptotic cells was observed at the highest dose. While this suggests that apoptosis can arise through alternative pathways, the absence of detectable DNA damage may reflect limitations in the sampling time and assay sensitivity, as DNA lesions measured by the comet assay can be transient and rapidly repaired.
References
Bajt, M. L., Ramachandran, A., Yan, H.-M., Lebofsky, M., Farhood, A., Lemasters, J. J. & Jaeschke, H. (2011). Apoptosis-Inducing Factor Modulates Mitochondrial Oxidant Stress in Acetaminophen Hepatotoxicity. Toxicological Sciences, 122(2), 598–605. https://doi.org/10.1093/toxsci/kfr116
Boon, N. J., Oliveira, R. A., Körner, P.-R., Kochavi, A., Mertens, S., Malka, Y., Voogd, R., Horst, S. E. M. van der, Huismans, M. A., Smabers, L. P., Draper, J. M., Wessels, L. F. A., Haahr, P., Roodhart, J. M. L., Schumacher, T. N. M., Snippert, H. J., Agami, R. & Brummelkamp, T. R. (2024). DNA damage induces p53-independent apoptosis through ribosome stalling. Science, 384(6697), 785–792. https://doi.org/10.1126/science.adh7950
Cao, Y., Sun, Q., Chen, Z., Lu, J., Geng, T., Ma, L. & Zhang, Y. (2022). CDKN2AIP is critical for spermiogenesis and germ cell development. Cell & Bioscience, 12(1), 136. https://doi.org/10.1186/s13578-022-00861-z
Chen, Y., Zhao, Y., Yang, X., Ren, X., Huang, S., Gong, S., Tan, X., Li, J., He, S., Li, Y., Hong, X., Li, Q., Ding, C., Fang, X., Ma, J. & Liu, N. (2022). USP44 regulates irradiation-induced DNA double-strand break repair and suppresses tumorigenesis in nasopharyngeal carcinoma. Nature Communications, 13(1), 501. https://doi.org/10.1038/s41467-022-28158-2
Cordelli, E., Leopardi, P., Villani, P., Marcon, F., Macrì, C., Caiola, S., Siniscalchi, E., Conti, L., Eleuteri, P., Malchiodi-Albedi, F. & Crebelli, R. (2010). Toxic and genotoxic effects of oral administration of furan in mouse liver. Mutagenesis, 25(3), 305–314. https://doi.org/10.1093/mutage/geq007
Elmore, S. (2007). Apoptosis: A Review of Programmed Cell Death. Toxicologic Pathology, 35(4), 495-516. doi:10.1080/01926230701320337
Habas, K., Anderson, D., & Brinkworth, M. (2016). Detection of phase specificity of in vivo germ cell mutagens in an in vitro germ cell system. Toxicology, 353-354, 1-10. doi:10.1016/j.tox.2016.04.001
Habas, K., Anderson, D., & Brinkworth, M. H. (2017). Germ cell responses to doxorubicin exposure in vitro. Toxicology Letters, 265, 70-76. doi:10.1016/j.toxlet.2016.11.016
Hamer, G., Roepers-Gajadien, H. L., Duyn-Goedhart, A. van, Gademan, I. S., Kal, H. B., Buul, P. P. W. van & Rooij, D. G. de. (2003). DNA double-strand breaks and gamma-H2AX signaling in the testis. Biology of Reproduction, 68(2), 628–634. https://doi.org/10.1095/biolreprod.102.008672
He, X., Zhu, L., Li, S., Chen, Z. & Zhao, X. (2018). Loperamide, an antidiarrheal agent, induces apoptosis and DNA damage in leukemia cells. Oncology Letters, 15(1), 765–774. https://doi.org/10.3892/ol.2017.7435
Jackson, S. P. & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078. https://doi.org/10.1038/nature08467
Kaina, B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochemical Pharmacology, 66(8), 1547–1554. https://doi.org/10.1016/s0006-2952(03)00510-0
Lee, J. (2005). ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science,308(5721), 551-554. doi:10.1126/science.1108297
Liu, W.-L., Gao, M., Tzen, K.-Y., Tsai, C.-L., Hsu, F.-M., Cheng, A.-L. & Cheng, J. C.-H. (2014). Targeting Phosphatidylinositide3-Kinase/Akt pathway by BKM120 for radiosensitization in hepatocellular carcinoma. Oncotarget, 5(11), 3662–3672. https://doi.org/10.18632/oncotarget.1978
Maréchal A, Zou L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harbor Perspectives in Biology. 2013;5(9):a012716. doi:10.1101/cshperspect.a012716.
Méniel, V., Megges, M., Young, M. A., Cole, A., Sansom, O. J., Clarke, A. R. (2015). Apc and p53 interaction in DNA damage and genomic instability in hepatocytes. Oncogene, 34(31), 4118–4129. https://doi.org/10.1038/onc.2014.342
Rodrigues, P. M. G., Grigaravicius, P., Remus, M., Cavalheiro, G. R., Gomes, A. L., Rocha-Martins, M., Martins, M. R., Frappart, L., Reuss, D., McKinnon, P. J., Deimling, A. von, Martins, R. A. P. & Frappart, P.-O. (2013). Nbn and Atm Cooperate in a Tissue and Developmental Stage-Specific Manner to Prevent Double Strand Breaks and Apoptosis in Developing Brain and Eye. PLoS ONE, 8(7), e69209. https://doi.org/10.1371/journal.pone.0069209
Roos, W., Baumgartner, M. & Kaina, B. (2004). Apoptosis triggered by DNA damage O6-methylguanine in human lymphocytes requires DNA replication and is mediated by p53 and Fas/CD95/Apo-1. Oncogene, 23(2), 359–367. https://doi.org/10.1038/sj.onc.1207080
Roos, W. P. & Kaina, B. (2006). DNA damage-induced cell death by apoptosis. Trends in Molecular Medicine, 12(9), 440–450. https://doi.org/10.1016/j.molmed.2006.07.007
Saha, S., Woodbine, L., Haines, J., Coster, M., Ricket, N., Barazzuol, L., Ainsbury, E., Sienkiewicz, Z. & Jeggo, P. (2014). Increased apoptosis and DNA double-strand breaks in the embryonic mouse brain in response to very low-dose X-rays but not 50 Hz magnetic fields. Journal of The Royal Society Interface, 11(100), 20140783. https://doi.org/10.1098/rsif.2014.0783
Suh, E.-K., Yang, A., Kettenbach, A., Bamberger, C., Michaelis, A. H., Zhu, Z., Elvin, J. A., Bronson, R. T., Crum, C. P. & McKeon, F. (2006). p63 protects the female germ line during meiotic arrest. Nature, 444(7119), 624–628. https://doi.org/10.1038/nature05337
Stringer, J. M., Winship, A., Zerafa, N., Wakefield, M. & Hutt, K. (2020). Oocytes can efficiently repair DNA double-strand breaks to restore genetic integrity and protect offspring health. Proceedings of the National Academy of Sciences,117(21), 11513–11522. https://doi.org/10.1073/pnas.2001124117
Talibova, G., Bilmez, Y. & Ozturk, S. (2022). DNA double-strand break repair in male germ cells during spermatogenesis and its association with male infertility development. DNA Repair, 118, 103386. https://doi.org/10.1016/j.dnarep.2022.103386
Tibbetts, R. S., Brumbaugh, K. M., Williams, J. M., Sarkaria, J. N., Cliby, W. A., Shieh, S.-Y., Taya, Y., Prives, C. & Abraham, R. T. (1999). A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & Development, 13(2), 152–157. https://doi.org/10.1101/gad.13.2.152
Urist, M., Tanaka, T., Poyurovsky, M. V. & Prives, C. (2004). p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes & Development, 18(24), 3041–3054. https://doi.org/10.1101/gad.1221004
Wang, W., Yuan, X., Mu, J., Zou, Y., Xu, L., Chen, J., Zhu, X., Li, B., Zeng, Z., Wu, X., Yin, Z. & Wang, Q. (2023). Quercetin induces MGMT+ glioblastoma cells apoptosis via dual inhibition of Wnt3a/β-Catenin and Akt/NF-κB signaling pathways. Phytomedicine, 118, 154933. https://doi.org/10.1016/j.phymed.2023.154933
Wawryk-Gawda, E., Chylińska-Wrzos, P., Lis-Sochocka, M., Chłapek, K., Bulak, K., Jędrych, M. & Jodłowska-Jędrych, B. (2014). P53 protein in proliferation, repair and apoptosis of cells. Protoplasma, 251(3), 525–533. https://doi.org/10.1007/s00709-013-0548-1
Williams, A. B. & Schumacher, B. (2016). p53 in the DNA-Damage-Repair Process. Cold Spring Harbor Perspectives in Medicine, 6(5), a026070. https://doi.org/10.1101/cshperspect.a026070
Xiao, H., Zhang, M., Wu, H., Wu, J., Hu, X., Pei, X., Li, D., Zhao, L., Hua, Q., Meng, B., Zhang, X., Peng, L., Cheng, X., Li, Z., Yang, W., Zhang, Q., Zhang, Y., Lu, Y. & Pan, Z. (2022). CIRKIL Exacerbates Cardiac Ischemia/Reperfusion Injury by Interacting With Ku70. Circulation Research, 130(5), e3–e17. https://doi.org/10.1161/circresaha.121.318992
Yauk, C. L., Buick, J. K., Williams, A., Swartz, C. D., Recio, L., Li, H., … Aubrecht, J. (2016). Application of the TGx‐28.65 transcriptomic biomarker to classify genotoxic and non‐genotoxic chemicals in human TK6 cells in the presence of rat liver S9. Environmental and Molecular Mutagenesis, 57(4), 243–260. http://doi.org/10.1002/em.22004
Zio, D. D., Cianfanelli, V. & Cecconi, F. (2012). New Insights into the Link Between DNA Damage and Apoptosis. Antioxidants & Redox Signaling, 19(6), 559–571. https://doi.org/10.1089/ars.2012.4938
Relationship: 3802: Apoptosis leads to Decrease, Sperm count
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Alkylation of DNA leading to decreased sperm count | adjacent | High | Low |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| Fetal | Low |
| Juvenile | High |
| Prepubertal | High |
| Adult, reproductively mature | High |
| Sex | Evidence |
|---|---|
| Male | High |
This KER is primarily supported by in vivo mammalian studies, particularly in rodents. The KER is most applicable when apoptosis is measured in early spermatogenesis, as apoptosis in late-stage germ cells plausibly affects sperm quality rather than sperm quantity. The sperm counts should be assessed following a delay consistent with the time required for the affected cells to progress through spermatogenesis. While the underlying biological processes are conserved across eukaryotic species that produce sperm, quantitative relationships may vary depending on species-specific spermatogenic kinetics.
Key Event Relationship Description
Apoptosis in germ cells directly depletes the developing germ cell population, whereas apoptosis in Sertoli cells and Leydig cells can indirectly reduce sperm counts by disrupting structural support and endocrine signaling in the testis that is required to sustain spermatogenesis.
Evidence Supporting this KER
Biological PlausibilityThe biological plausibility of this KER is strong, based on extensive mechanistic understanding of spermatogenesis and the role of apoptosis in regulating testicular cell populations.
Spermatogenesis is a continuous and highly coordinated process of germ cell proliferation and differentiation, leading to the production of mature spermatozoa. Apoptosis (programmed cell death) is an important mechanism that removes damaged cells and maintains tissue homeostasis (Shaha et al., 2010). During spermatogenesis, physiologic apoptosis occurs in all testicular cells (Oldereid et al., 2001). Meanwhile, stress-induced apoptosis can be triggered by exogenous stressors, such as environmental toxicants, heat, and radiation, which can markedly increase apoptotic cell death in testis (Shaha et al., 2010).
Mechanistically, the primary link between apoptosis and reduced sperm counts is the loss of developing male germ cells, which decreases the number of cells available to complete spermatogenesis and produce mature spermatozoa. However, apoptosis of testicular Sertoli and Leydig cells can also contribute to reduced sperm counts through indirectmechanisms, such as disrupting structural support and hormonal regulation of spermatogenesis.
Apoptosis in male germ cells
In the early stage of spermatogenesis, spermatogonial stem cells (SSCs) maintain the male germline through self-renewal, while a subset of SSCs commits to differentiation to produce mature sperm (Nakagawa et al., 2010). As long-term germ cell reservoirs, SSCs have enhanced DNA repair capacity, which renders them less sensitive to damage or stress-induced apoptosis compared with their differentiating progeny (Meistrich et al., 1982b; Rübe et al., 2011). However, excessive apoptosis of SSCs at higher doses can compromise the ability of the testis to replenish the pool of developing germ cells and cause sustained or irreversible impairment of spermatogenesis (Meistrich et al., 2013).
Differentiating spermatogonia are particularly sensitive to stress-induced apoptosis because they undergo rapid mitotic divisions (Meistrich et al., 1982a, 1982b, 2013). Apoptosis of differentiating spermatogonia results in a progressive loss of the more mature germ cells in a process known as maturation depletion (Meistrich et al., 2013). Although later-stage germ cells continue to develop, minimal replacement occurs from progenitor cells. Consequently, the pool of developing germ cells is depleted over time, leading to reductions in sperm counts that become evident after a delay. This delay reflects the time required for developing germ cells to complete spermatogenesis before the effects of progenitor cell depletion become apparent in mature sperm output. As a result, the timing of the decline in sperm counts is generally consistent with the duration of spermatogenesis in the species of interest (Meistrich et al., 1992).
Primary spermatocytes undergo extensive meiotic reorganization and division to generate haploid spermatids. Disruption of meiotic progression induces checkpoint mediated apoptosis to eliminate defective cells, particularly at the pachytene stage (Roeder and Bailis, 2000; Li et al., 2025). Compared with pre-meiotic germ cells, apoptosis in spermatocytes is often detected during meiotic progression rather than as an immediate response following exposure (Meistrich et al., 1982b, 2013). The subsequent reduction in sperm counts is usually temporary when SSCs survive, as these remaining stem cells rapidly replenish spermatogenic cells and restore sperm counts in ~45 days in mice and ~12 weeks in humans (Meistrich et al., 2013).
After meiosis, spermatids differentiate into mature spermatozoa during spermiogenesis. At this stage, male germ cells undergo chromatin and morphological remodeling when the sperm genome becomes highly condensed and transcriptionally silenced (Aitken et al., 2010). In parallel, mitochondria are relocalized, either becoming confined to the sperm midpiece or transferred into residual bodies for degradation and elimination (Varuzhanyan and Chan, 2020). These structural changes limit the execution of canonical caspase-dependent apoptotic pathways. Instead, post-meiotic germ cells undergo truncated or abortive apoptosis, which is mediated primarily by reactive oxygen species and lipid peroxidation; the formation of lipid adducts leads to a rapid loss of sperm motility within hours, reflecting immediate functional loss (Aitken and Baker, 2013). Notably, these cells remain viable, and this form of apoptosis does not typically result in immediate DNA fragmentation and will not be detected by the TUNEL assay (Aitken and Baker, 2013). Accordingly, compared with apoptosis in early-stage germ cells, apoptosis occurring at post-meiotic stages is less likely to cause substantial germ cell loss and reduction in sperm counts despite causing functional defects.
Apoptosis in other testicular cells
In addition to germ cells, apoptosis can also happen in other testicular cells and indirectly influence the progression of spermatogenesis. Sertoli cells provide essential structural support in the seminiferous epithelium and play multiple roles in maintaining testicular functions, including supporting germ cell development, facilitating germ cell movement across the epithelium, enabling spermiation (release of mature spermatids), and secreting regulatory factors (Murphy and Richburg, 2015). As one Sertoli cell only has a finite supportive capacity, the size of Sertoli cell population is a key determinant of the total germ cell numbers and, indirectly, Leydig cell numbers in the testis (Rebourcet et al., 2017). Under physiological conditions, spontaneous apoptosis is an important mechanism to maintain the Sertoli cell to germ cell ratio. Toxicant-induced Sertoli cell apoptosis has been reported to cause germ cell loss through loss of structural and metabolic support, ultimately leading to reduced sperm output (Murphy and Richburg, 2015). Among germ cell populations, the developing spermatocytes are the most sensitive to Sertoli cell-selective apoptosis, while spermatogonia are relatively resistant (Murphy and Richburg, 2015). Leydig cells are responsible for testosterone synthesis, which is essential for multiple key processes during spermatogenesis, including germ cell survival and progression (Smith and Walker, 2014). Hormonal deprivation significantly increases the incidence of apoptotic germ cells, particularly late-stage spermatocytes that are androgen-dependent (Hikim et al., 1997). Excessive apoptosis in Leydig cells is strongly linked to lower intratesticular testosterone levels, and therefore can impair germ cell survival, indirectly leading to reduced sperm counts.
Together, these well-established mechanisms provide strong biological support for a causal relationship between increased apoptosis in testicular cells and reduced sperm counts.
Empirical EvidenceThe empirical support for this KER is strong. Concordant increases in apoptosis in testicular cells and reductions in sperm counts have been consistently observed across multiple in vivo rodent studies, and numerous intervention studies provide evidence of essentiality by demonstrating preservation or recovery of sperm counts following attenuation of apoptotic signaling. Temporal concordance is strongly supported by the well-established kinetics of spermatogenesis, although it is often inferred rather than directly measured within individual studies. Quantitative dose-response information is limited because many studies employ single exposure or intervention conditions. In addition, apoptosis frequently co-occurs with oxidative stress and inflammatory responses, which may contribute to spermatogenic impairment and complicate attribution to apoptosis alone. Nevertheless, these limitations do not detract from the overall consistency, biological coherence, and weight of evidence supporting this KER.
A large number of studies demonstrated the use of chemicals with anti-apoptotic, anti-inflammatory, and anti-oxidant properties to attenuate toxicant-induced testicular injury. These studies consistently show that attenuation of apoptosis is associated with recovery of sperm counts, providing supportive, albeit indirect, evidence for the essentiality of apoptosis. Since this is a data-rich field and the biological plausibility is strong, select empirical studies are presented as examples.
In a study by Yaman et al (2018), 30 day-old male Wistar albino rats were exposed to a single intraperitoneal dose of 5 mg/kg cisplatin. Half of the rats were euthanized at 31 days of age (pre-pubertal) and observed for the presence of testicular cell apoptosis; the other half was euthanized at 90 days (fertile age) to evaluate epididymal sperm counts. In exposed pre-pubertal rats, 107 ± 5.9 apoptotic (TUNEL-positive) cells were observed in 100 seminiferous tubule cross-sections, compared with only 24.6 ± 6.1 cells in control rats (p<0.001). The TUNEL+ cells were predominantlyspermatogonia and spermatocytes. Consistent with the expected temporal sequent, the average sperm counts measuredlater in adulthood were significantly reduced by ~54% (175 ± 67.1 million/mL in treated rats compared to 379.2 ± 45.9 million/mL observed in the controls) (Figure 1). Moreover, co-treatment with L-carnitine (250 mg/kg, for three consecutive days) significantly decreased the numbers of TUNEL+ cells in the tubules and resulted in a near-complete restoration of sperm counts (Figure 1). This study provides strong evidence of temporal concordance and indirect evidence of essentiality, as modulation of apoptosis was associated with recovery of sperm production.
Udefa et al. (2020) reported preventive effects of a tigernut plant extract on lead acetate-induced testicular damage in rats. Male Wistar rats were injected intraperitoneally with 20 mg/kg lead acetate once daily, with or without oral co-treatment with the extract (500 or 1000 mg/kg) for three weeks. Lead exposure significantly increased apoptosis in the testes, evidenced by pro-apoptotic shift in the Bax/Bcl-2 ratio (~7-fold increase) and elevated cleaved caspase-3 levels (~2.3-fold increase), indicating activation of pro-apoptotic pathways. Concordant decreases in serum and testicular testosterone levels suggested toxicity to Leydig cells. These changes were accompanied by a ~32% reduction in epididymal sperm counts. Co-treatment with the plant extract reversed changes in apoptotic markers and restored sperm counts in a dose-dependent manner. A negative correlation (r= -0.813, p<0.05) between the Bax/Bcl-2 ratio and sperm counts was reported, based on a small number of treatment group-level data. Since the Bax/Bcl-2 ratio is an indirect marker of apoptotic signaling, this correlation provides only limited quantitative support for a direct response-response relationship between the two KEs.
Oyovwi et al. (2023) investigated whether quercetin, a bioflavanol known for anti-oxidant and anti-apoptotic properties, could mitigate testicular dysfunction as a side effect of the antiepileptic drug levetiracetam (LEV). Male Wistar rats (10-12 weeks old) were exposed to saline or LEV (300 mg/kg/day) by oral gavage for 56 days, a period that covers a full cycle of spermatogenesis in rats. A separate group of rats were co-treated with quercetin (20 mg/kg/day) during days 28-56, administered 30-minute after each LEV dose. LEV exposure altered apoptosis-related factors in testicular tissues, including decreased expression of anti-apoptotic protein Bcl-2, and increased pro-apoptotic signaling. Specifically, the cytochrome c level in mitochondrial supernatant was increased by ~60%, while expression of caspase-3 and p53, and sperm DNA fragmentation index (indirectly predicted by aniline blue staining) showed about 2 to 3-fold increases relative to controls (Figure 2). These changes were accompanied by lower testicular weights, a ~30% decrease in epididymal sperm counts, and histological evidence of germ cell loss at all stages, along with decreases in Sertoli cell and Leydig cell numbers. Co-treatment with quercetin reversed all changes in apoptotic markers to control levels and prevented the LEV-induced reductions in testicular weights and sperm counts. This study provides high-quality evidence supporting essentiality of apoptosis in male germ cells.
In a mouse study examining the effects of X-ray radiation on male infertility, selenium nanoparticles (SeNPs) and a probiotic Lactobacillus casei were shown to mitigate testicular damage through anti-apoptotic and anti-oxidant mechanisms (Ehghaghi et al., 2022). Sixty-four Syrian male mice were divided into eight groups (n=8 per group) and orally treated with vehicle (PBS), SeNPs (0.2 mg/kg/day), probiotic (1 × 10⁸ CPU), or their combination. Four groups received whole body X-ray radiation (2 Gray for 5 minutes) after each daily oral dosing for 30 days, and the other four groups were used as non-irradiated controls. Apoptosis of testicular cells was assessed using Annexin V and propidium iodide (PI) double staining and flow cytometry. In the absence of irradiation, none of the groups exhibited significant changes in testicular parameters (Figure 3). X-ray exposure caused a ~20-fold increase in late apoptotic testicular cells, alongside a ~65% decrease in spermatogonia cell numbers and > 50% decrease in testicular sperm counts (Figure 3). Treatment with either SeNPs or the probiotic reduced apoptotic signaling, as indicated by downregulation of caspase-3 and caspase-9 transcripts. These changes were accompanied by partial restoration of spermatogonia and sperm counts. The combined treatment restored both apoptotic markers and downstream endpoints to near-control levels.
In addition to the select studies presented above, multiple rodent studies have reported chemo-preventive effects of anti-apoptotic interventions and corresponding recovery of impaired spermatogenesis caused by a wide range of stressors, including high-fat diet (Abdulwahab et al., 2021), nanomaterials (reviewed by Sun et al., 2025), and toxicants such as aflatoxin B1 (Ijaz et al., 2023), tartrazine (Essawy et al., 2024), atrazine (reviewed by Abarikwu et al., 2023), suggesting a broad applicability of this KER. Mitigation of testicular cells apoptosis is consistently concordant with preservation of sperm counts.
Together, these findings demonstrate concordant changes in apoptosis and sperm counts across treatment groups and provide indirect evidence of essentiality, as attenuation of apoptosis (and other stress responses) was associated with recovery of sperm production. Although evidence for dose-response relationships is limited, the studies generally support dose and temporal alignment and a moderate empirical relationship for this KER.
Uncertainties and InconsistenciesA key uncertainty arises from the use of whole testis homogenates to measure apoptotic markers, which reflect mixed cell populations and do not distinguish responses of germ cells and somatic cells. Histological evaluation of seminiferous tubules supports germ cell loss as a primary driver of reduced sperm counts (Yaman et al., 2018; Udefa et al, 2020; Ehghaghi et al., 2022; Oyovwi et al., 2023; Ijaz et al., 2023). However, reductions in Sertoli cell and Leydig cell numbers can occur in parallel (Oyovwi et al., 2023). Therefore, the reduced sperm output is likely caused by combined effects on mixed populations of testicular cells and cumulative damages over the course of spermatogenesis, although the relative contribution of each cell type is not clear.
An apparently inconsistent study was noted. In a rat study, exposure to 10 mg/kg of sodium arsenite for 14 days induced a near 100% increase in the mRNA expression of Bax and caspase-3, and a ~40% decrease in Bcl-2 expression in testicular tissues, indicating activation of apoptotic signaling. Although histological analysis showed remarkable loss of spermatogenic cells, particularly spermatogonia, no changes were observed in testicular weights or epididymal sperm counts. Suppression of apoptotic signaling by carvacrol treatment did not alter sperm density across groups (Gur et al., 2023). The inconsistency likely reflects a limitation of the study design rather than a lack of biological linkage between apoptosis and sperm counts. Samples were collected immediately following the 14-day exposure period , which is insufficient to observe downstream changes in sperm output given the duration of spermatogenesis in rats.
Multiple biological pathways can influence sperm output and introduce variability and uncertainty in this KER.Inflammation and oxidative stress frequently co-occur with apoptosis, and therefore reductions in sperm output may reflect overlapping or combined effects of these stress-related responses. In many studies, attenuation of apoptosis is often accompanied by simultaneous suppression of oxidative stress and inflammatory markers (Udefa et al., 2020; Oyovwi et al, 2023). Given the extensive interplay among stress responsive pathways, it is difficult to separate their individual contributions. In addition, disruption of hormonal regulation in the hypothalamic-pituitary-gonadal axis can indirectly affect spermatogenesis and sperm production. Such interacting mechanisms may partly explain inconsistencies observed across studies and should be considered when interpreting this KER.
References
Abarikwu, S. O., Ezim, O. E., Ikeji, C. N. & Farombi, E. O. (2023). Atrazine: cytotoxicity, oxidative stress, apoptosis, testicular effects and chemopreventive Interventions. Frontiers in Toxicology, 5, 1246708. https://doi.org/10.3389/ftox.2023.1246708
Abdulwahab, D. K., Ibrahim, W. W., El-Aal, R. A. A., Abdel-Latif, H. A. & Abdelkader, N. F. (2021). Grape seed extract improved the fertility-enhancing effect of atorvastatin in high-fat diet-induced testicular injury in rats: involvement of antioxidant and anti-apoptotic effects. Journal of Pharmacy and Pharmacology, 73(3), 366–376. https://doi.org/10.1093/jpp/rgaa002
Aitken, R. J. & Baker, M. A. (2013). Causes and consequences of apoptosis in spermatozoa; contributions to infertility and impacts on development. International Journal of Developmental Biology, 57(2-3–4), 265–272. https://doi.org/10.1387/ijdb.130146ja
Aitken, R. J., Findlay, J. K., Hutt, K. J. & Kerr, J. B. (2010). Apoptosis in the germ line. Reproduction, 141(2), 139–150. https://doi.org/10.1530/rep-10-0232
Ehghaghi, A., Zokaei, E., Modarressi, M. H., Tavoosidana, G., Ghafouri-Fard, S., Khanali, F., Motevaseli, E. & Noroozi, Z. (2022). Antioxidant and anti-apoptotic effects of selenium nanoparticles and Lactobacillus casei on mice testis after X-ray. Andrologia, 54(11), e14591. https://doi.org/10.1111/and.14591
Essawy, A., Matar, S., Mohamed, N., Abdel-Wahab, W. & Abdou, H. (2024). Ginkgo biloba extract protects against tartrazine-induced testicular toxicity in rats: involvement of antioxidant, anti-inflammatory, and anti-apoptotic mechanisms. Environmental Science and Pollution Research, 31(10), 15065–15077. https://doi.org/10.1007/s11356-024-32047-0
Gur, C., Akarsu, S. A., Akaras, N., Tuncer, S. C. & Kandemir, F. M. (2023). Carvacrol reduces abnormal and dead sperm counts by attenuating sodium arsenite-induced oxidative stress, inflammation, apoptosis, and autophagy in the testicular tissues of rats. Environmental Toxicology, 38(6), 1265–1276. https://doi.org/10.1002/tox.23762
Hikim, A. P. S., Rajavashisth, T. B., Hikim, I. S., Lue, Y., Bonavera, J. J., Leung, A., Wang, C. & Swerdloff, R. S. (1997). Significance of apoptosis in the temporal and stage-specific loss of germ cells in the adult rat after gonadotropin deprivation. Biology of Reproduction, 57(5), 1193–1201. https://doi.org/10.1095/biolreprod57.5.1193
Antioxidant, anti-inflammatory, and anti-apoptotic effects of genkwanin against aflatoxin B1-induced testicular toxicity. Toxicology and Applied Pharmacology, 481, 116750. https://doi.org/10.1016/j.taap.2023.116750
Meistrich, M. L. (1982a). Quantitative Correlation Between Testicular Stem Cell Survival, Sperm Production, and Fertility in the Mouse After Treatment With Different Cytotoxic Agents. Journal of Andrology, 3(1), 58–68. https://doi.org/10.1002/j.1939-4640.1982.tb00646.x
Meistrich, M. L., Finch, M., Cunha, M. F. da, Hacker, U. & Au, W. W. (1982b). Damaging effects of fourteen chemotherapeutic drugs on mouse testis cells. Cancer Research, 42(1), 122–131.
Meistrich, M. L., Wilson, G., Brown, B. W., Cunha, M. F. da & Lipshultz, L. I. (1992). Impact of cyclophosphamide on long-term reduction in sperm count in men treated with combination chemotherapy for Ewing and soft tissue sarcomas. Cancer, 70(11), 2703–2712. https://doi.org/10.1002/1097-0142(19921201)70:11<2703::aid-cncr2820701123>3.0.co;2-x
Murphy, C. J. & Richburg, J. H. (2015). Implications of Sertoli cell induced germ cell apoptosis to testicular pathology. Spermatogenesis, 4(2), e979110. https://doi.org/10.4161/21565562.2014.979110
Nakagawa, T., Sharma, M., Nabeshima, Y., Braun, R. E. & Yoshida, S. (2010). Functional Hierarchy and Reversibility Within the Murine Spermatogenic Stem Cell Compartment. Science, 328(5974), 62–67. https://doi.org/10.1126/science.1182868
Oldereid, N. B., Angelis, P. D., Wiger, R. & Clausen, O. P. F. (2001). Expression of Bcl-2 family proteins and spontaneous apoptosis in normal human testis*. Molecular Human Reproduction, 7(5), 403–408. https://doi.org/10.1093/molehr/7.5.403
Oyovwi, M. O., Oghenetega, O. B., Victor, E., Faith, F. Y. & Uchechukwu, J. G. (2023). Quercetin protects against levetiracetam induced gonadotoxicity in rats. Toxicology, 491, 153518. https://doi.org/10.1016/j.tox.2023.153518
Rebourcet, D., Darbey, A., Monteiro, A., Soffientini, U., Tsai, Y. T., Handel, I., Pitetti, J.-L., Nef, S., Smith, L. B. & O’Shaughnessy, P. J. (2017). Sertoli Cell Number Defines and Predicts Germ and Leydig Cell Population Sizes in the Adult Mouse Testis. Endocrinology, 158(9), 2955–2969. https://doi.org/10.1210/en.2017-00196
Rübe, C. E., Zhang, S., Miebach, N., Fricke, A. & Rübe, C. (2011). Protecting the heritable genome: DNA damage response mechanisms in spermatogonial stem cells. DNA Repair, 10(2), 159–168. https://doi.org/10.1016/j.dnarep.2010.10.007
Shaha, C., Tripathi, R. & Mishra, D. P. (2010). Male germ cell apoptosis: regulation and biology. Philosophical Transactions of the Royal Society B: Biological Sciences, 365(1546), 1501–1515. https://doi.org/10.1098/rstb.2009.0124
Smith, L. B. & Walker, W. H. (2014). The regulation of spermatogenesis by androgens. Seminars in Cell & Developmental Biology, 30, 2–13. https://doi.org/10.1016/j.semcdb.2014.02.012
Sun, Y., Zhou, Y., Xie, D., Wang, X., Wang, Y., Liang, Y. & Luo, X. (2025). Preclinical Evaluation of Protective Effects of Terpenoids Against Nanomaterial-Induced Toxicity: A Meta-Analysis. Journal of Applied Toxicology, 45(7), 1080–1102. https://doi.org/10.1002/jat.4716
Udefa, A. L., Amama, E. A., Archibong, E. A., Nwangwa, J. N., Adama, S., Inyang, V. U., Inyaka, G. U., Aju, G. J., Okpa, S. & Inah, I. O. (2020). Antioxidant, anti-inflammatory and anti-apoptotic effects of hydro-ethanolic extract of Cyperus esculentus L. (tigernut) on lead acetate-induced testicular dysfunction in Wistar rats. Biomedicine & Pharmacotherapy, 129, 110491. https://doi.org/10.1016/j.biopha.2020.110491
Varuzhanyan, G. & Chan, D. C. (2020). Mitochondrial dynamics during spermatogenesis. Journal of Cell Science, 133(14), jcs235937. https://doi.org/10.1242/jcs.235937
Yaman, O., & Topcu-Tarladacalisir, Y. (2018). L-carnitine counteracts prepubertal exposure to cisplatin induced impaired sperm in adult rats by preventing germ cell apoptosis. Biotechnic & Histochemistry, 1-11. doi:10.1080/10520295.2017.1401661
List of Non Adjacent Key Event Relationships
Relationship: 3771: Alkylation, DNA leads to Decrease, Sperm count
AOPs Referencing Relationship
| AOP Name | Adjacency | Weight of Evidence | Quantitative Understanding |
|---|---|---|---|
| Alkylation of DNA leading to decreased sperm count | non-adjacent | High | Low |
Evidence Supporting Applicability of this Relationship
| Life Stage | Evidence |
|---|---|
| All life stages |
| Sex | Evidence |
|---|---|
| Male | High |
This KER is supported primarily by mammalian data (rodents and humans) and is applicable to other eukaryotic species that produce sperm. DNA adducts can occur in any cell type, but this KER is specific to the male germline. Although reduced sperm count can only be measured after sexual maturity, DNA alkylation may occur during fetal, juvenile, or adult life stages. Exposure during early development can impair germ cell populations and lead to delayed reductions in sperm production that become evident after reproductive maturation.
Key Event Relationship Description
DNA alkylating agents can form covalent adducts in the DNA of any exposed cell types; however, actively proliferating germ cells, particularly spermatogonia, are especially susceptible to the cytotoxic consequences of alkylation damage. During DNA replication, alkyl adducts can interfere with replication fork progression and activate DNA damage response pathways. Persistent or unrepaired lesions can result in cell cycle arrest and apoptosis, leading to depletion of differentiating germ cell populations and, consequently, reduced sperm production.
Evidence Supporting this KER
Biological PlausibilityThe biological plausibility of this KER is strong. DNA alkylation refers to the covalent addition of alkyl groups (e.g., methyl, ethyl, propyl) to nucleotides at noncanonical positions in DNA, generating lesions that interfere with DNA replication, transcription, and chromosome integrity (Soll et al., 2017). These lesions can occur across all stages of spermatogenesis and trigger downstream signalling cascades that lead to apoptosis. Across species, actively proliferating cells have long been shown to be particularly susceptible to chemotherapeutic alkylating drugs due to ongoing DNA replication; this provides a conserved biological basis for alkylation-induced impairment of spermatogenesis (Drewinko et al., 1981; Meistrich, 1982a).
Unrepaired alkylation damage in male germ cells can block DNA replication and induce base mispairing and strand breaks, which activate DNA damage response pathways and lead to cell cycle arrest and selective elimination of damaged cells through programmed cell death (Kaina, 2003). This stringent quality control mechanism is important to preserve germline genomic integrity; however, it inevitably reduces the supply of cells progressing through spermatogenesis (Agarwal et al., 2020; Li et al., 2025). Therefore, DNA alkylation-induced apoptosis in germ cells can plausibly lead to reduced sperm counts.
The consequences of alkylation-induced apoptosis on sperm counts vary depending on the developmental stage at which the damage occurs. Alkylation of DNA in proliferating or differentiating germ cells at early (pre-meiotic) stages compromises the ability of the testis to replenish the pool of developing germ cells and can cause sustained or irreversible reductions in sperm counts (Meistrich et al., 2013).
Meiosis gives rise to spermatocytes, which have tightly regulated meiotic DNA damage checkpoints to coordinate repair of endogenous programmed DNA strand breaks (Hunter, 2015). Alkylation-induced DNA lesions in meiotic cells can increase the overall repair burden and interfere with the tightly regulated repair of programmed meiotic breaks. When lesions persist, checkpoint signaling promotes the elimination of defective meiotic cells (Roeder and Bailis, 2000; Li et al., 2025). However, this effect is typically temporary because surviving SSCs can repopulate the seminiferous epithelium over time (Meistrich, 1982a).
In contrast, post-meiotic germ cells in late spermatogenesis are largely DNA repair deficient; thus, DNA damage at this stage can only be repaired after fertilization through maternal repair mechanisms (Olsen et al., 2005; Newman et al., 2021). Alkylation of DNA in post-meiotic germ cells does not typically drive germ cell death (and thus reduced sperm counts) contributing instead to sperm DNA damage and reduced sperm quality.
In addition to germ cells, alkylation damage in testicular Sertoli and Leydig cells may also indirectly contribute to reduced sperm counts through disruption of structural support and hormonal regulation required for spermatogenesis. However, differentiating germ cells are considered particularly sensitive targets of alkylating agents due to ongoing DNA replication, and are likely the primary drivers of alkylation-induced impairment of sperm production.
In addition to impacts on adult spermatogenesis, clinical and experimental studies demonstrate that exposure to alkylating chemotherapeutic agents during prepubertal or adolescent ages can impair germ cell populations and lead to persistent reductions in sperm production detectable in adulthood (reviewed by Delessard et al., 2019). Alterations in Sertoli cells and Leydig cells have also been reported following chemotherapy exposure, albeit the effects are inconsistent across studies and do not yet allow definitive conclusions (Delessard et al., 2019). These observations in cancer survivors and experimental models support the relevance of early-life stages and the delayed manifestation of impaired spermatogenesis following chemotherapy.
Together, these well-established biological processes support strong plausibility for a causal relationship between DNA alkylation damage and reduced sperm counts. While alkylation damage can occur across all stages of spermatogenesis, effects on sperm counts are primarily driven by damage to proliferating and meiotic germ cells and have typically been observed following exposure in sexually mature individuals. However, exposures during early developmental stages may result in more persistent or severe impairments due to disruption of the germ cell pool, with effects becoming apparent later in adulthood.
Empirical EvidenceStrong empirical evidence is available from experimental animal models and human clinical observations. Although direct measurement of both DNA adducts and sperm counts in the same study is limited, a substantial body of evidence demonstrates consistent patterns of temporal and dose concordance across studies examining well-characterized DNA alkylating agents. Human data further support the biological relevance of this relationship.
Altakroni et al. (2021) analyzed 105 semen samples from men undergoing assisted reproduction treatment (ART)and quantified the levels of N7-methyldeoxyguanosine (N7-MedG), a biomarker of exposure to methylating agents, in sperm genomic DNA using an immunoslotblot assay. A negative correlation was noted between N7-MedG concentrations and sperm concentration (Spearman's rank correlation coefficient= -0.32, p<0.001). Logistic regression analyses demonstrated that the elevated N7-MedG level was significantly associated with low sperm concentration (odds ratio [OR] = 6.46, 95% CI: 1.51-27.7); this association remained strong after adjustment for other DNA damage markers, including the percentage of sperm with high DNA damage and median % tail DNA measured by the neutral comet assay (adjusted OR = 6.42, 95% CI: 1.47-28.0) (Altakroni et al., 2021). This study provides evidence linking DNA alkylation to reduced sperm counts in humans.
It is well characterized that various DNA adducts rapidly form in mouse male germ cells within two hours after exposure to alkylating agents: N-ethyl-N-nitrosourea (ENU), ethyl methanesulfonate (EMS), and diethyl sulfate (DES). The frequency of adduct formation increases with dose, supporting dose-dependent activation of the upstream KE. Data for ENU exposure are shown as examples (Figure 1) (van Zeeland et al., 1990).
Accordingly, DNA adduct formation can be reasonably inferred following exposures to well-characterized DNA alkylating agents. Empirical evidence supporting this KER primally comes from studies in animals and humans. Below, we summarize human clinical observations and selected data on several alkylating medications as examples to illustrate consistent patterns of reduced sperm counts following exposure.
Evidence in human
Azoospermia (absence of sperm in semen, < 0.1 million cells/mL) and oligozoospermia (semen with a low sperm count, 0.1-10 million cells/mL) are well-recognized side effects of cancer treatment with alkylating agents in men of reproductive age (reviewed by Howell and Shalet, 2005; Okada and Fujisawa, 2018). Dramatic declines in ejaculated sperm counts can occur within 1-2 months after receiving the treatment, while azoospermia usually occur after 2 months (Meistrich et al., 2013). The duration and reversibility of treatment-induced azoospermia is largely dependent on the dose of the drugs administered (Okada and Fujisawa, 2018).
Clinical studies provide evidence linking treatment with alkylating agents to reduced sperm counts in humans. More than 50% of patients with malignant lymphomas developed azoospermia after receiving chlorambucil treatment at a cumulative dose of more than 400 mg (Richter et al., 1970; Cheviakoff et al., 1973). Similarly, in 35 patients treated with cyclophosphamide, sperm production in 22 patients declined to azoospermic levels, and 6 declined to oligozoospermic levels within a few months after treatment initiation (Meistrich et al., 1992). In this cohort, patients received less than 7.5 g/m2 (median dose 4.1 g/m2) of cyclophosphamide restored sperm production to a normal range within 5 years, whereas only 10% of patients recovered when treated with higher doses (> 7.5 g/m2), likely reflecting SSC depletion due to cumulative cytotoxicity (Meistrich et al., 1992).
Consistent patterns have also been reported for chemotherapy regimens containing alkylating agents. For instance, MVPP (mustine, vinblastine, procarbazine, and prednisolone) and COPP (cyclophosphamide, vincristine, procarbazine, and prednisolone) caused azoospermia in more than 90% of adult patients (reviewed by Howell and Shalet, 2005). When the doses of alkylating agents were reduced in these regimens, gonadotoxicity was also reduced, suggesting the role of DNA alkylation in causing reduced sperm counts and a dose-dependent relationship (Okada and Fujisawa, 2018).
Long-term reproductive effects of alkylating drugs and irradiation during early life stages have been reported. Green et al. (2014) evaluated semen parameters in 214 adult male survivors of childhood cancer who had received alkylating chemotherapeutic drugs. Azoospermia was diagnosed in 53 (25%) of participants and oligospermia in 59 (28%); the mean cumulative alkylating agent dose was higher in both groups than in participants with normospermia. Moreover, alkylating agent exposure was negatively correlated with sperm concentrations (r=-0.37, p<0.0001); specifically, each 1000 mg/m2 increase in cumulative dose increased the odds of azoospermia by 22% (Green et al., 2014).
Beaud et al. (2019) revealed correlations between sperm parameters and treatment characteristics in 13 childhood cancer survivors. The participants received various doses of Mustargen®, procarbazine, or cyclophosphamide around puberty. Despite the small sample size, a strong link was observed between lower sperm counts and the use of alkylating agents (Spearman's rank correlation coefficient r=-0.62, p≤0.05), but not the use of other two non-alkylating drug classes, vinca alkaloids (r=0.07, p>0.05) and anthracyclines (r=-0.02, p>0.05) (Beaud et al., 2019).
Evidence in animal models in vivo
Meistrich (1982a) examined the effects of triethylenethiophosphoramide (thio-TEPA) on testicular cell survival and sperm production in C3H mice. Male mice were injected i.p. with eight doses of thio-TEPA (0-35 mg/kg), and the number of sperm heads was counted in testis homogenates. Thio-TEPA exerts its anticancer activity through the formation of alkyl DNA adducts (Musser et al., 1989; Grigorii et al., 1991). A clear dose-dependent decrease in sperm head numbers was observed 8 weeks post-treatment. At doses ≥ 10 mg/kg, sperm counts were reduced to less than 10% of the control level, while the highest dose (35 mg/kg) reduced the number to ~1% of control (Meistrich, 1982a). These findings demonstrate dose concordance, with increasing exposure to this known alkylating agents associated with progressively greater reductions in sperm counts, and these decreases occur subsequent to treatment with alkylating agents, consistent with temporal concordance.
The same research group further examined spermatogenesis in C3H mice following single i.p. injections of 14 chemotherapeutic drugs, including 7 alkylating agents (Meistrich et al., 1982b). Eleven days post-treatment, histological analyses showed that spermatogonial cells were the most sensitive germ cell population to cell death, whereas the spermatocytes and spermatids were relatively resistant at this time point, consistent with known biology. Dose-dependent loss of differentiating spermatogonia was observed following treatment with lomustine (40 mg/kg), mustargen (3 mg/kg), carmustine (10 and 60 mg/kg), mitomycin C (1.5 and 5 mg/kg), chlorambucil (20 mg/kg), thio-TEPA (3 and 26 mg/kg), and procarbazine (120 and 800 mg/kg). Assessment of sperm production at later time points supports temporal concordance between alkylation-induced germ cell loss and downstream reductions in sperm counts. Testicular sperm counts in testis homogenates measured 29 days post-treatment confirmed moderate to strong killing effects of alkylating agents on differentiating spermatogonia; all drugs reduced sperm counts to less than 10% of control within the dose range tested. Sperm counts measured 56 days after treatment reflect stem cell survival and recovery capacity of sperm production. At this timepoint, lomustine and mustargen induced up to a 25% decrease in sperm head numbers, whereas carmustine, chlorambucil, mitomycin C, and procarbazine reduced sperm counts to 30-60% of control levels. Nearly no sperm were counted in testes treated with thio-TEPA at 26 mg/kg, indicating minimal stem cell survival and recovery. Thus, a large number of alkylating agents lead to spermatogonial cell death and decline in sperm counts.
Busulfan, a potent bifunctional alkylating agent, produced similar effects. Bucci and Meistrich (1987) reported clear dose-dependent decreases in testicular sperm counts in C3H mice exposed to busulfan (6, 13, 20, 28, 40 mg/kg) via i.p. injection. The dose required to reduce sperm head numbers by 50% of controls was estimated at 10 mg/kg at 29 days and 8 mg/kg at 56 days post-treatment. At 44 weeks, testicular and epididymal sperm counts remained significantly decreased by more than 50% in mice treated with busulfan ≥ 20 mg/kg compared to controls, while sperm counts returned to normal ranges in the lower dose groups (Bucci and Meistrich, 1987). These data demonstrate delayed onset and persistence of reduced sperm counts following exposure to a potent alkylating agent, supporting strong dose and temporal concordance and limited recovery of SSCs at higher doses.
Similar findings have been reported in a non-human primate model, supporting the conservation of this KER across species. Hermann et al. (2009) reported the effects of a single intravenous exposure to busulfan (4, 8, 12 mg/kg) exposure on spermatogenesis in adult rhesus macaques. Semen samples were collected weekly and total sperm counts per ejaculate were monitored for up to 52 weeks. Exposure to all three doses of busulfan caused dose-dependent and progressive decreases in sperm counts, which declined to zero by 7 weeks in animals treated with 12 mg/kg busulfan, and by 10 weeks following treatment with 4 mg/kg and 8 mg/kg busulfan. Testis volumes were also decreased correspondingly. Animals treated with 4 mg/kg busulfan restored normal spermatogenesis by 24 weeks following the exposure, albeit the sperm count is still lower than those in the controls. In contrast, sperm counts remained undetectable 52 weeks following exposure to 8 mg/kg and 12 mg/kg busulfan, suggesting SSC depletion and a longe-term loss of fertility. This study provides strong evidence for temporal and dose concordance, as reductions in sperm counts occur after exposure with a delay consistent with spermatogenic progression, with higher doses leading to more rapid and sustained depletion of sperm.
Repeated administration of a potent alkylating agent, ENU (100 mg/kg/week for three weeks), in C57BL/6J mice caused a delayed but significant reduction in sperm counts (Yin et al., 2014). No significant change in sperm count was observed during the first two weeks following the initial exposure, confirming that lack of apoptotic response in post-meiotic germ cells; however, sperm counts declined markedly from weeks 3-8 and reached near-zero levels by week 8. Sperm counts then gradually recovered, reaching approximately 85% of control levels by week 12, indicating profound but reversible impairment of spermatogenesis following ENU exposure (Figure 2). This study provides indirect support for essentiality through a stop-start (stressor removal) design.
In another Balb/c mouse study examining germline chromatin damage and sperm counts, ENU was used as a positive control. A single i.p. injection of 75 mg/kg of ENU at 8 weeks of age resulted in a delayed ~43% decrease in cauda epididymal sperm counts at 18 weeks of age, relative to control, accompanied by increased sperm DNA fragmentation and elevated germline mutation frequencies (Swayne et al., 2012). These studies provide strong temporal evidence.
Prenatal exposure studies provide evidence that early life exposure to alkylating agents and ionizing radiation can cause long-term effects on spermatogenesis. Comish et al. (2014) exposed pregnant 129 mice to either whole-body irradiated (0.8 Gy) or cyclophosphamide (7.5 mg/kg, i.p.) on embryonic days E10.5 and E11.5, corresponding to a critical period of primordial germ cell development. Male offspring were evaluated for testicular development and sperm production. At 4 weeks of age, male offspring from both treatment groups presented significantly reduced testicular weights (~70% of control), indicative of significant germ cell loss. The average number of atrophic Sertoli cell-only seminiferous tubules increased from 1.7 per testis section in controls to 4.8-5.8 tubules per section in irradiated and cyclophosphamide-treated animals. At 8 weeks of age, testicular weight remained reduced to 78% of control values, while testicular head counts and epididymal sperm counts were reduced to 62%-70% of control levels. These findings demonstrate that alkylation damage during fetal life can result in long-lasting germ cell depletion and reductions in sperm production in adulthood.
Together, these findings demonstrate that alkylation-induced genomic damage in male germ cells is consistently associated with impaired spermatogenesis and reduced sperm output. Although direct measurement of DNA adduct formation and sperm counts within the same study is uncommon, parallel evidence from studies of well-characterized alkylating agents shows consistent, dose-dependent changes in both KEs, supporting dose concordance. Significant reductions in sperm output are typically observed weeks to months following exposure, reflecting the time required for species-specific spermatogenic transit and confirming temporal concordance. The extent, duration, and reversibility of these effects highly depend on exposure dose, survival of SSCs, and spermatogenic kinetics in the test species.
Uncertainties and InconsistenciesSeveral uncertainties remain in this KER. First, DNA alkyl adducts in germ cells and sperm output are rarely measured in the same study, and many studies rely on surrogate markers of DNA damage, such as DNA strand breaks, making it difficult to attribute downstream effects specifically to DNA alkylation.
Second, reduced sperm count can arise through multiple mechanisms. In addition to direct alkylation-induced DNA damage in germ cells, other defects such as spermiation failure (i.e., retention of mature spermatozoa in late stage tubules) (Meistrich et al., 1982b), Sertoli cell dysfunction (Cui et al. 2024), and hormonal disruption (Sriram et al., 2024) may co-occur with alkylation damage and contribute to reduced testicular or epididymal sperm output. Moreover, alkylating agents have been shown to induce epigenetic alterations in sperm, including changes in DNA methylation at specific fertility-related genes (Altakroni et al., 2021). These alterations are associated with sperm DNA damage and reduced sperm counts (Jenkins et al., 2016); however, reported correlations between DNA damage markers and methylation are gene-specific and inconsistent across studies (Altakroni et al., 2021). As such, the relative contribution of epigenetic dysregulation to reduced sperm counts in this context remains uncertain.
Although Altakroni et al. (2021) reported an association between N7-MedG levels and decreased sperm concentration in 105 patients, this result was not replicated in a smaller follow-up study (n=16) (Altakroni et al., 2025). The latter study was based on a limited number of samples with N7-MedG measurements, which may have reduced statistical power. No significant correlations between N7-MedG levels, sperm concentration, or neutral comet assay measures (% tail DNA), despite observing a significant association with reduced fertilization rates.
References
Agarwal, A., Majzoub, A., Baskaran, S., Selvam, M. K. P., Cho, C. L., Henkel, R., Finelli, R., Leisegang, K., Sengupta, P., Barbarosie, C., Parekh, N., Alves, M. G., Ko, E., Arafa, M., Tadros, N., Ramasamy, R., Kavoussi, P., Ambar, R., Kuchakulla, M., … Shah, R. (2020). Sperm DNA Fragmentation: A New Guideline for Clinicians. The World Journal of Men’s Health, 38(4), 412–471. https://doi.org/10.5534/wjmh.200128
Altakroni, B., Nevin, C., Carroll, M., Murgatroyd, C., Horne, G., Brison, D. R. & Povey, A. C. (2021). The marker of alkyl DNA base damage, N7-methylguanine, is associated with semen quality in men. Scientific Reports, 11(1), 3121. https://doi.org/10.1038/s41598-021-81674-x
Altakroni, B., Hunter, H., Horne, G., Brison, D. R. & Povey, A. C. (2025). DNA damage in prepared semen is negatively associated with semen quality and fertilisation rate in assisted reproduction technology (ART) treatment. Human Fertility,28(1), 2442450. https://doi.org/10.1080/14647273.2024.2442450
Beaud, H., Albert, O., Robaire, B., Rousseau, M. C., Chan, P. T. K. & Delbes, G. (2019). Sperm DNA integrity in adult survivors of paediatric leukemia and lymphoma: A pilot study on the impact of age and type of treatment. PLoS ONE, 14(12), e0226262. https://doi.org/10.1371/journal.pone.0226262
Bucci, L. R. & Meistrich, M. L. (1987). Effects of busulfan on murine spermatogenesis: cytotoxicity, sterility, sperm abnormalities, and dominant lethal mutations. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 176(2), 259–268. https://doi.org/10.1016/0027-5107(87)90057-1
Comish, P. B., Drumond, A. L., Kinnell, H. L., Anderson, R. A., Matin, A., Meistrich, M. L. & Shetty, G. (2014). Fetal Cyclophosphamide Exposure Induces Testicular Cancer and Reduced Spermatogenesis and Ovarian Follicle Numbers in Mice. PLoS ONE, 9(4), e93311. https://doi.org/10.1371/journal.pone.0093311
Cheviakoff, S., Calamera, J. C., Morgenfeld, M. & Mancini, R. E. (1973). Recovery of spermatogenesis in patients with lymphoma after treatment with chlorambucil. Reproduction, 33(1), 155–157. https://doi.org/10.1530/jrf.0.0330155
Cui, Y., Harteveld, F., Omar, H. A. M. B., Yang, Y., Bjarnason, R., Romerius, P., Sundin, M., Nyström, U. N., Langenskiöld, C., Vogt, H., Henningsohn, L., Frisk, P., Vepsäläinen, K.,
Delessard, M., Saulnier, J., Rives, A., Dumont, L., Rondanino, C. & Rives, N. (2020). Exposure to Chemotherapy During Childhood or Adulthood and Consequences on Spermatogenesis and Male Fertility. International Journal of Molecular Sciences, 21(4), 1454. https://doi.org/10.3390/ijms21041454
Green, D. M., Kawashima, T., Stovall, M., Leisenring, W., Sklar, C. A., Mertens, A. C., Donaldson, S. S., Byrne, J. & Robison, L. L. (2009). Fertility of Male Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. Journal of Clinical Oncology, 28(2), 332–339. https://doi.org/10.1200/jco.2009.24.9037
Grigorii, V. A., Leonid, F. S., Tatyana, L. P., Oleg, A. B., Olga, Yu. L. & Vadim, S. S. (1991). Direct observation of the alkylation products of deoxyguanosine and DNA by fast atom bombardment mass spectrometry. Biological Mass Spectrometry, 20(11), 665–668. https://doi.org/10.1002/bms.1200201103
Musser, S. M., Pan, S. S. & Callery, P. S. (1989). Liquid chromatography-thermospray mass spectrometry of DNA adducts formed with mitomycin C, porfiromycin and thiotepa. Journal of Chromatography, 474(1), 197–207. https://doi.org/10.1016/s0021-9673(01)93915-9
Petersen, C., Mitchell, R. T., Guo, J., Alves-Lopes, J. P., Jahnukainen, K. & Stukenborg, J.-B. (2024). Prior exposure to alkylating agents negatively impacts testicular organoid formation in cells obtained from childhood cancer patients. Human Reproduction Open, 2024(3), hoae049. https://doi.org/10.1093/hropen/hoae049
Drewinko, B., Patchen, M., Yang, L. Y. & Barlogie, B. (1981). Differential killing efficacy of twenty antitumor drugs on proliferating and nonproliferating human tumor cells. Cancer Research, 41(6), 2328–2333.
Hermann, B. P., Sukhwani, M., Lin, C., Sheng, Y., Tomko, J., Rodriguez, M., Shuttleworth, J. J., McFarland, D., Hobbs, R. M., Pandolfi, P. P., Schatten, G. P. & Orwig, K. E. (2009). Characterization, Cryopreservation, and Ablation of Spermatogonial Stem Cells in Adult Rhesus Macaques. Stem Cells, 25(9), 2330–2338. https://doi.org/10.1634/stemcells.2007-0143
Howell, S. J. & Shalet, S. M. (2005). Spermatogenesis After Cancer Treatment: Damage and Recovery. JNCI Monographs, 2005(34), 12–17. https://doi.org/10.1093/jncimonographs/lgi003
Hunter, N. (2015). Meiotic Recombination: The Essence of Heredity. Cold Spring Harbor Perspectives in Biology, 7(12), a016618. https://doi.org/10.1101/cshperspect.a016618
Jenkins, T. G., Aston, K. I., Hotaling, J. M., Shamsi, M. B., Simon, L. & Carrell, D. T. (2016). Teratozoospermia and asthenozoospermia are associated with specific epigenetic signatures. Andrology, 4(5), 843–849. https://doi.org/10.1111/andr.12231
Kaina, B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochemical Pharmacology, 66(8), 1547–1554. https://doi.org/10.1016/s0006-2952(03)00510-0
Li, N., Wang, H., zou, S., Yu, X. & Li, J. (2025). Perspective in the Mechanisms for Repairing Sperm DNA Damage. Reproductive Sciences, 32(1), 41–51. https://doi.org/10.1007/s43032-024-01714-5
Meistrich, M. L. (1982a). Quantitative Correlation Between Testicular Stem Cell Survival, Sperm Production, and Fertility in the Mouse After Treatment With Different Cytotoxic Agents. Journal of Andrology, 3(1), 58–68. https://doi.org/10.1002/j.1939-4640.1982.tb00646.x
Meistrich, M. L., Finch, M., Cunha, M. F. da, Hacker, U. & Au, W. W. (1982b). Damaging effects of fourteen chemotherapeutic drugs on mouse testis cells. Cancer Research, 42(1), 122–131.
Meistrich, M. L., Wilson, G., Brown, B. W., Cunha, M. F. da & Lipshultz, L. I. (1992). Impact of cyclophosphamide on long-term reduction in sperm count in men treated with combination chemotherapy for Ewing and soft tissue sarcomas. Cancer, 70(11), 2703–2712. https://doi.org/10.1002/1097-0142(19921201)70:11<2703::aid-cncr2820701123>3.0.co;2-x
Newman, H., Catt, S., Vining, B., Vollenhoven, B. & Horta, F. (2021). DNA repair and response to sperm DNA damage in oocytes and embryos, and the potential consequences in ART: a systematic review. Molecular Human Reproduction, 28(1), gaab071. https://doi.org/10.1093/molehr/gaab071
Okada, K. & Fujisawa, M. (2018). Recovery of Spermatogenesis Following Cancer Treatment with Cytotoxic Chemotherapy and Radiotherapy. The World Journal of Men’s Health, 36(2), 166–174. https://doi.org/10.5534/wjmh.180043
Olsen, A.-K., Lindeman, B., Wiger, R., Duale, N. & Brunborg, G. (2005). How do male germ cells handle DNA damage? Toxicology and Applied Pharmacology, 207(2), 521–531. https://doi.org/10.1016/j.taap.2005.01.060
Richter, P., Calamera, J. C., Morgenfeld, M. C., Kierszenbaum, A. L., Lavieri, J. C. & Mancini, R. E. (1970). Effect of chlorambucil on spermatogenesis in the human with malignant lymphoma. Cancer, 25(5), 1026–1030. https://doi.org/10.1002/1097-0142(197005)25:5<1026::aid-cncr2820250506>3.0.co;2-c
Roeder, G. S. & Bailis, J. M. (2000). The pachytene checkpoint. Trends in Genetics : TIG, 16(9), 395–403. https://doi.org/10.1016/s0168-9525(00)02080-1
Rübe, C. E., Zhang, S., Miebach, N., Fricke, A. & Rübe, C. (2011). Protecting the heritable genome: DNA damage response mechanisms in spermatogonial stem cells. DNA Repair, 10(2), 159–168. https://doi.org/10.1016/j.dnarep.2010.10.007
Soll, J. M., Sobol, R. W. & Mosammaparast, N. (2017). Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends in Biochemical Sciences, 42(3), 206–218. https://doi.org/10.1016/j.tibs.2016.10.001
Stocks, S. J., Agius, R. M., Cooley, N., Harrison, K. L., Brison, D. R., Horne, G., Gibbs, A. & Povey, A. C. (2010). Alkylation of sperm DNA is associated with male factor infertility and a reduction in the proportion of oocytes fertilised during assisted reproduction. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 698(1–2), 18–23. https://doi.org/10.1016/j.mrgentox.2010.02.019
Sriram, S., Macedo, T., Mavinkurve-Groothuis, A., Wetering, M. van de & Looijenga, L. H. J. (2024). Alkylating agents-induced gonadotoxicity in prepubertal males: Insights on the clinical and preclinical front. Clinical and Translational Science, 17(7), e13866. https://doi.org/10.1111/cts.13866
Swayne, B. G., Kawata, A., Behan, N. A., Williams, A., Wade, M. G., MacFarlane, A. J. & Yauk, C. L. (2012). Investigating the effects of dietary folic acid on sperm count, DNA damage and mutation in Balb/c mice. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 737(1–2), 1–7. https://doi.org/10.1016/j.mrfmmm.2012.07.002
van Zeeland, A. A., Groot, A. de & Neuhäuser-Klaus, A. (1990). DNA adduct formation in mouse testis by ethylating agents: a comparison with germ-cell mutagenesis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 231(1), 55–62. https://doi.org/10.1016/0027-5107(90)90176-5
Yin, J., Sun, K. & Chen, B. (2014). Time-dependent toxic effects of N-ethyl-N-nitrosourea on the testes of male C57BL/6J mice: a histological and ultrastructural study. International Journal of Clinical and Experimental Pathology, 8(2), 1830–1843.